Mutagenic Impurity Risk Assessment Throughout the Development and Manufacturing Process
Since the early 2000s and the advent of the first guidance relating to mutagenic impurities developed by the European Medicines Agency (EMA), it has been necessary to assess the risk posed by mutagenic impurities. Although initially it was thought that avoidance was an option, it quickly became apparent that this was not a viable strategy. The current state of chemical methodology is such that it is impossible to construct synthetic molecules without the use of reagents that are either potentially mutagenic or mutagenic; that these are required to form carbon bonds. Having established this, it then becomes a question of risk assessment and establishment of effective control.
ICH M7 is unique among ICH guidelines in that it is applicable throughout development, as opposed to others which may be only applicable at the time of marketing application. As a consequence, a mutagenic impurity risk assessment is required prior to any clinical evaluation of the drug in question. So how is this done? ICH M7 provides a very good overview of what is required. However, it is important to establish a systematic approach to precisely conduct the risk assessment in practice.
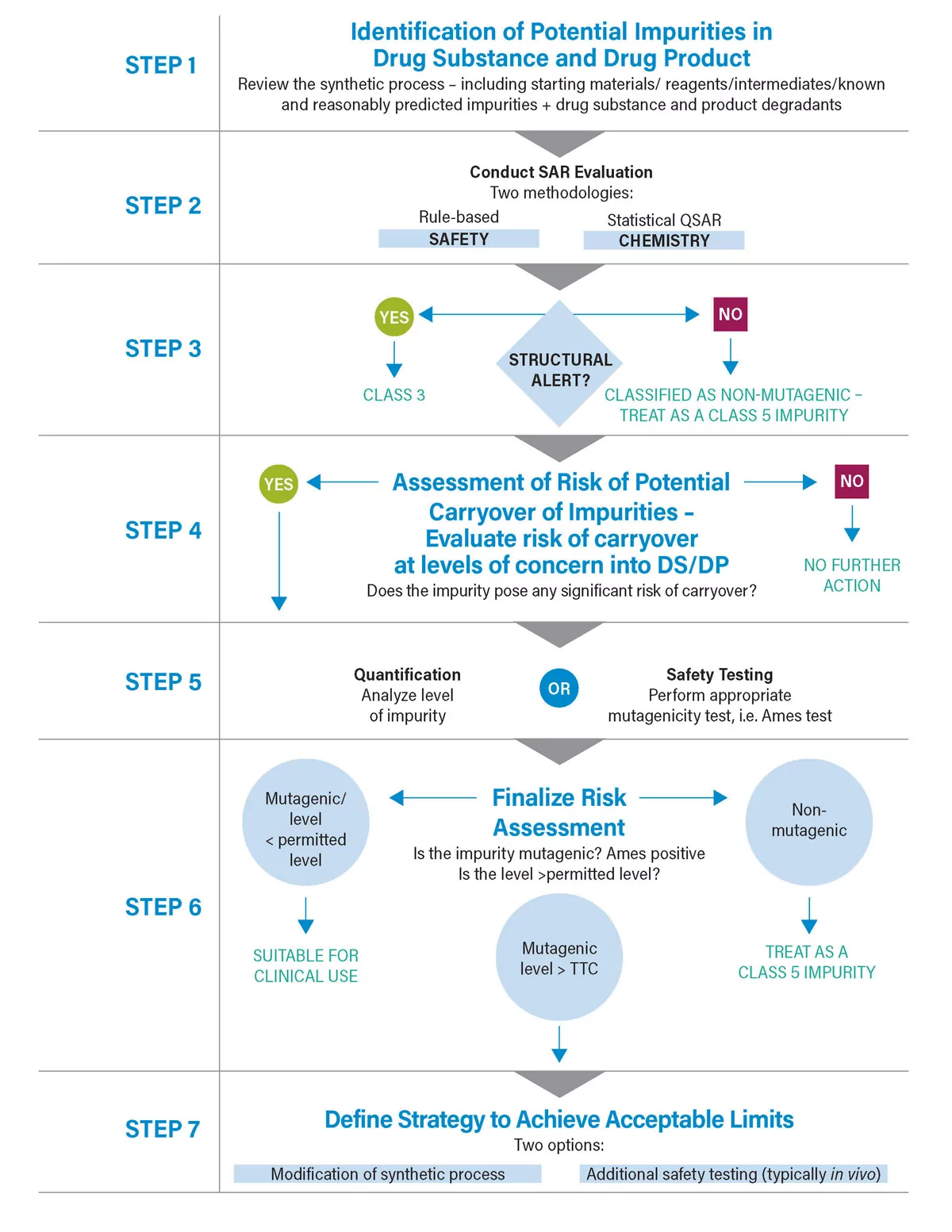
Outlined below is a process flow diagram that describes one potential approach. It is important to note this requires a multi-disciplinary approach. In many ways the process can be thought of as a relay; once one aspect is completed it is handed over to another discipline. i.e. from chemistry to safety, or chemistry to analysis until the risk assessment is complete. In its simplest term, the risk assessment needs to answer 2 questions:
- Are there any mutagenic impurities associated with the manufacture and storage of the drug (both active and formulated product)? If yes then:
- Are they present at levels of concern?
It is only if the answer to question 2 is “yes” that there is an issue.
Briefly examine each step. Perhaps the most difficult is Step 1: where in the process and what impurities should be assessed? ICH M7 states that this should include starting materials, reagents, intermediates, identified and reasonably predicted impurities. Of these, the most challenging is the last. How do you define whether an impurity is reasonably predicted all possible impurities? Recent issues relating to N Nitrosamines bring this into sharp focus. This was a case of unexpected, non-predicted, and potentially cancer-causing impurity that came about in manufacturing and ending up in multiple drug products. What is needed is a good understanding of potential side reactions that may generate mutagenic impurities. Attempts are currently underway to ensure a better understanding in this vital area.
Once relevant impurities have been identified, the next step is to assess the potential mutagenicity of the impurities concerned. i.e. SAR (Structure Activity Relationship) evaluation. ICH M7 is quite prescriptive, insisting on the use of in -silico tools to do this and specifying the use of two different methodologies. It also stresses the importance of expert knowledge to assess results. Such an approach is possible because of the strong correlation between structure and mutagenicity.
SAR evaluation effectively filters out close to 90% of impurities, leaving a small number of either mutagenic or potentially mutagenic compounds. The next stage is to further assess risk. We do this by assessing the actual mutagenicity of the impurity — typically through a reverse mutation bacterial assay – or by evaluating the potential for carryover at levels of concern, either through calculation or analysis.
If needed, ICH M7 includes a number of control options. Option 4 allows control to be demonstrated through assessing the potential for carryover by evaluating the physico-chemical properties of the impurity and correlating these to the process conditions (Purge Factor). If this provides insufficient assurance, testing may be required. Such trace analysis can be challenging, given the low levels involved and the often highly reactive nature of the impurity.
Once all the data are collected, the risk assessment can be completed and the critical second question answered (Do the mutagenic impurities exist at levels of concern?). Experience shows that using a systematic approach such as that described usually demonstrates control of the impurity. My next blog digs deeper into the process set out by global regulatory agencies to quantify and evaluate the presence of N-nitrosamines. Stay tuned!
Andy Teasdale, PhD is a Senior Principal Scientist, Impurity Management and External Advocacy at AstraZeneca
Related posts
OligoWorks SPE Kits: Boosting Oligonucleotide Bioanalysis Efficiency
New OligoWorks Solid Phase Extraction (SPE) Kits for extraction and bioanalysis of oligonucleotides deliver advantages over liquid-liquid extraction (LLE) and other existing SPE kits and sample preparation methods. Read on to discover how.

Increasing Lab Efficiency with Amazon Web Services to Enable Real-Time Remote Access to Lab Information
The collaboration between Waters and AWS has enhanced the waters_connect System Monitoring tool, revolutionizing laboratory productivity. This cloud-based platform provides secure, compliant access to chromatography lab information, allowing users to coordinate work, address system errors, and make informed decisions from anywhere, anytime.

Embracing Sustainability in Analytical Science
The integration of sustainability practices in analytical laboratories is not just a trend but a crucial shift towards a more responsible future. The ACQUITY QDa II Mass Detector shows how Waters enhances lab capabilities while also promoting sustainability.
Popular Topics
ACQUITY QDa (17) bioanalysis (11) biologics (14) biopharma (26) biopharmaceutical (36) biotherapeutics (17) case study (17) chromatography (14) data integrity (22) food analysis (12) HPLC (15) LC-MS (22) liquid chromatography (LC) (20) mass detection (16) mass spectrometry (MS) (54) method development (13) STEM (12) sustainability (12)