通过打破孤岛来提高分析生产力(第一部分)
从上游到下游的实验室
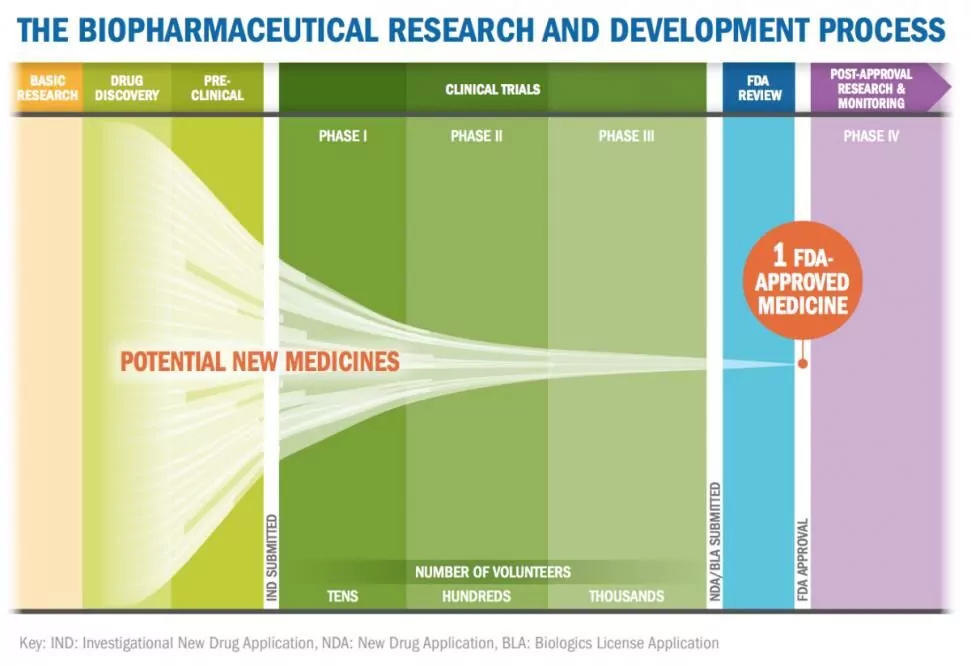
药品的发现和开发过程通常以简化的图形形式表示,例如以下。

以这种方式来看,它似乎是一种直接的线性努力,分子通过一系列的步骤单向移动 - 但实际上并非如此。通常情况下,分子会向下游推进,但随后会出现分析问题(例如:数据工件或新发现的杂质),分子将被送回上游进行进一步的分析和开发,然后再次恢复其下游之旅。
此外,如果你看一下沿途每个阶段正在进行的候选药物的数量,并应用一个时间表,整个过程被更准确地描绘成一个漏斗,如图所示。
当你考虑到许多最初在上游调查的候选药物在开发途径的不同阶段被淘汰,以及一个分子平均需要10到15年才能完成成为FDA批准的药物的旅程时,你就可以理解今天开发一个新药的平均成本(包括失败)是如何落在8亿到10亿美元之间的。
再加上不断变化的监管要求变得越来越严格(尤其是对大型和日益复杂的生物制药),以及在减少开发和生产成本的同时加快上市速度的压力越来越大(由激烈的竞争、快速兴起的生物仿制药市场和寻求控制医疗成本的政府推动),很明显,生物制药行业需要实现更高水平的生产力。
为此,今天经常发生重大生产力损失的一个领域是在实验室和地域之间转移数据和方法的过程中,特别是当从上游实验室转移到受监管的下游实验室(后期开发、制造和质量控制)时,方法和系统验证、数据完整性和合规性至关重要。
正如Waters最近的一篇博文《今天是什么改变了分析方法的转移?第三部分:跨越国界的方法,一家大型全球制药公司的科学家估计,一种分析方法在其生命周期内可能会有多达100次的转移。 人们通常不理解的是,在转移数据和方法时,尤其是在转移到下游受监管的实验室以及在这些实验室之间转移时,隐藏着许多复杂的问题和风险。所使用的仪器平台和软件往往随着你在药物发现和开发管道中的穿越而变化。因此,方法必须经常被重新开发和重新验证,而历史数据和方法调整的理由可能会被有效地困在难以进入的分析仓中。
如果处理不当,合规性和数据完整性问题很可能被监管机构标记出来,这反过来又会导致大量增加的工作和成本超支的重大延误。
Unger咨询公司的Barbara Unger在她最近的文章 "An Analysis of FDA FY2017 Drug GMP Warning Letters"中指出,2017年FDA向药品生产基地发出的警告信数量增加了一倍。其中大部分与合规性和数据完整性问题有关,包括方法和系统验证、数据获取和存储、审计跟踪等。
随着生物制药行业的不断扩大,以及越来越多的产品进入市场,包括生物仿制药和生物代用品,证明分析合规性和数据完整性的挑战只会变得更加突出。 为了提高开发速度和生产力,公司必须有一个明确的全组织战略,以打破分析孤岛,满足合规性和数据完整性要求。
在沃特世,我们正在努力创建一个单一的、符合合规要求的信息学框架,我们所有适合的工具以及第三方工具都可以在这个框架中运行;这个框架可以使数据和信息从上游实验室到下游实验室、从中心实验室到分布式实验室以及在所有地区顺利流动;这个框架可以保持数据的完整性和合规性,创造工作流程的效率,促进生产力的大幅提升。
在我的下一篇博客中,我将讨论与外部合作伙伴合作以及打破中央和分布式实验室之间的孤岛的挑战。
访问 waters.com/tamethechaos 欲了解更多信息。
以前关于生物制药数据协调的博文。
相关帖子
OligoWorks SPE Kits: Boosting Oligonucleotide Bioanalysis Efficiency
New OligoWorks Solid Phase Extraction (SPE) Kits for extraction and bioanalysis of oligonucleotides deliver advantages over liquid-liquid extraction (LLE) and other existing SPE kits and sample preparation methods. Read on to discover how.

Increasing Lab Efficiency with Amazon Web Services to Enable Real-Time Remote Access to Lab Information
The collaboration between Waters and AWS has enhanced the waters_connect System Monitoring tool, revolutionizing laboratory productivity. This cloud-based platform provides secure, compliant access to chromatography lab information, allowing users to coordinate work, address system errors, and make informed decisions from anywhere, anytime.

将可持续性融入分析科学
将可持续发展实践融入分析实验室不仅是一种趋势,而且是迈向更负责任的未来的关键转变。ACQUITY QDa II 质量检测器展示了沃特世如何在增强实验室能力的同时促进可持续发展。
热门话题
ACQUITY QDa (17) bioanalysis (11) biologics (14) biopharma (26) biopharmaceutical (36) biotherapeutics (17) case study (17) chromatography (14) data integrity (22) food analysis (12) HPLC (15) LC-MS (22) liquid chromatography (LC) (20) mass detection (16) mass spectrometry (MS) (54) method development (13) STEM (12) sustainability (12)