Fatty acids, both free and as part of complex lipids, play a number of key roles in metabolism – as major metabolic fuel (storage and transport of energy), as essential components of all membranes, and as gene regulators. In addition, dietary lipids provide polyunsaturated fatty acids that are precursors of powerful locally acting metabolites, e.g., eicosanoids.
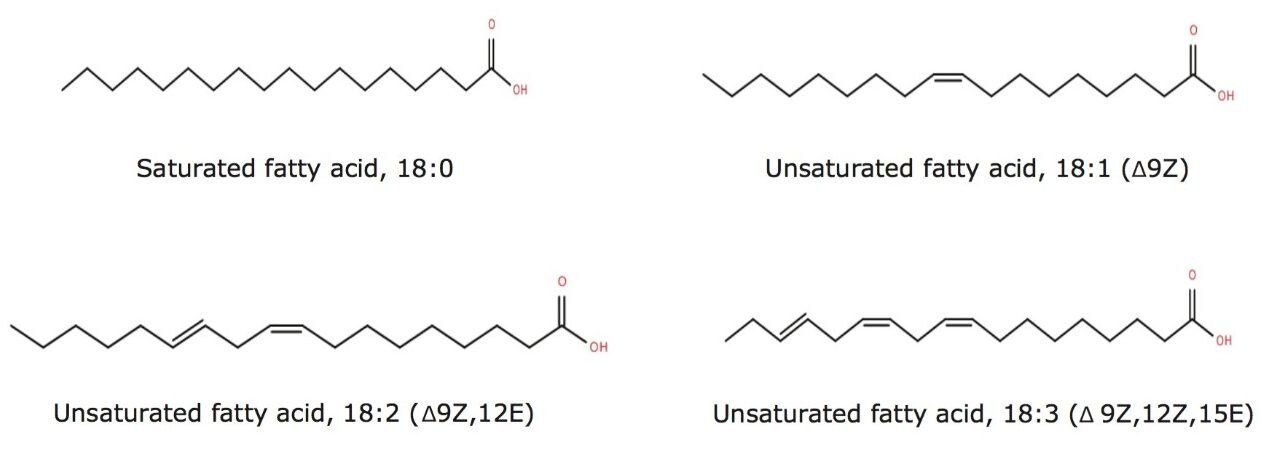
The common fatty acids of animal and plant origin have even-numbered chains of 16 to 24 carbon atoms with 0 to 6 double bonds. Nature provides countless exceptions, however, and odd- and even-numbered fatty acids with up to nearly 100 carbon atoms exist. In addition, double bonds can be of the cis (Z) and trans (E) configuration and there can be innumerable other structural features, including branch points, rings, oxygenated functions, and many more.
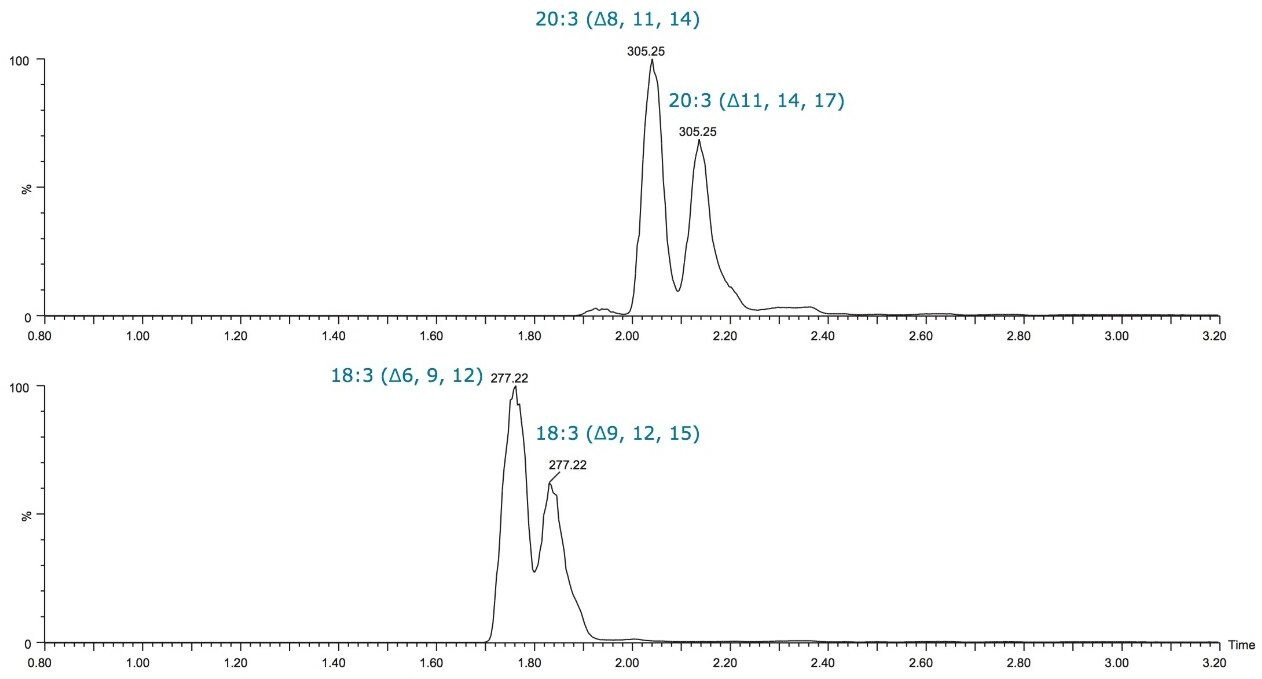
Fatty acid chains may contain one or more double bonds at specific positions (unsaturated and poly unsaturated with cis (Z) or trans (E) configuration) or they may be fully saturated. The LIPIDMAPS systematic nomenclature for fatty acids indicates the location of double bonds with reference to the carboxyl group with “Δ”.1 Fatty acid structures also contain a methyl group at one end of the molecule (designated omega, ω) and a carboxyl group at the other end. The carbon atom next to the carboxyl group is called α carbon and the subsequent one the β carbon. The letter “n” is also often used instead of ω to indicate the position of the double bond closest to the methyl end.2 Figure 1 outlines the structures of different straight chain fatty acids.
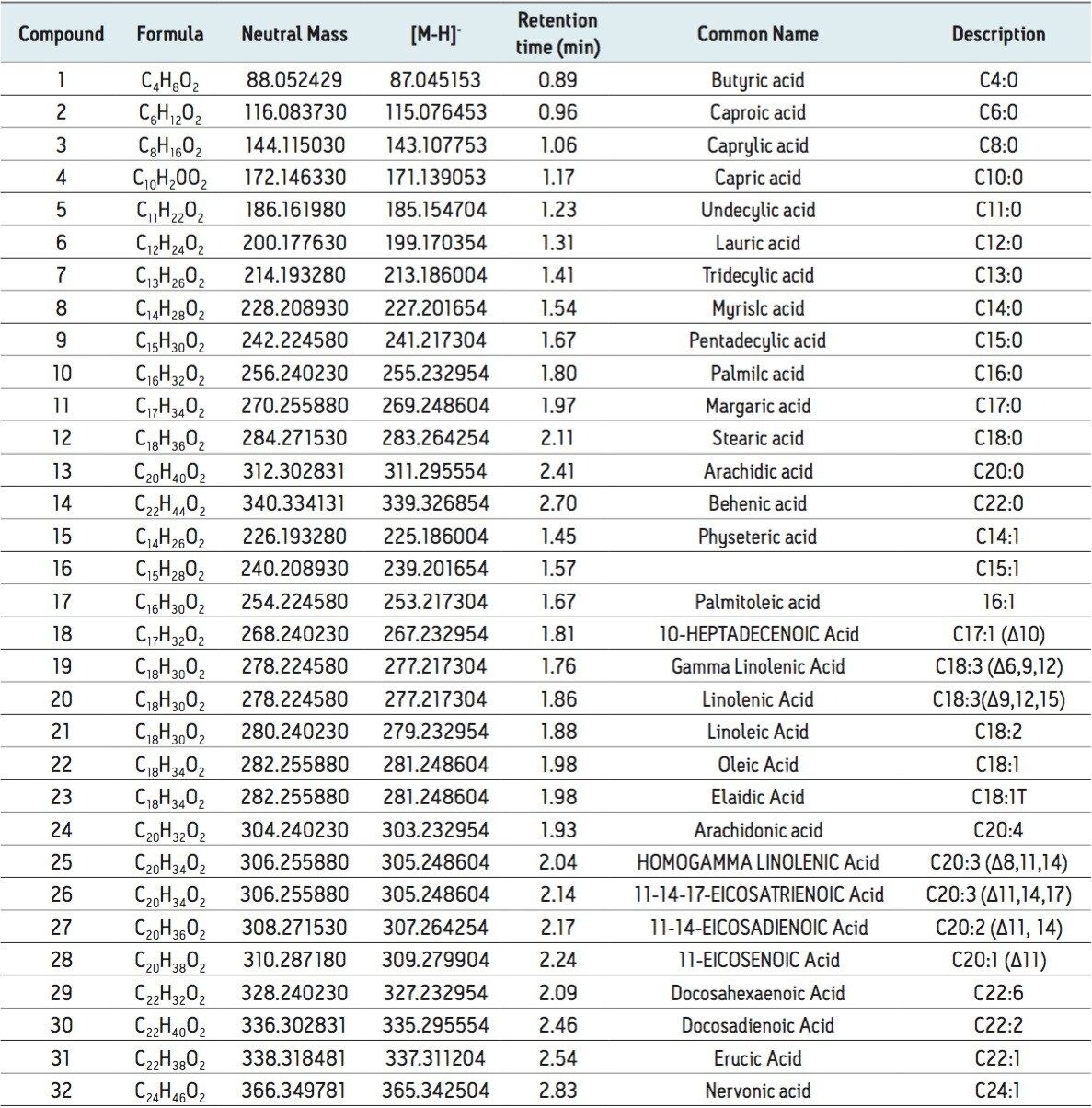
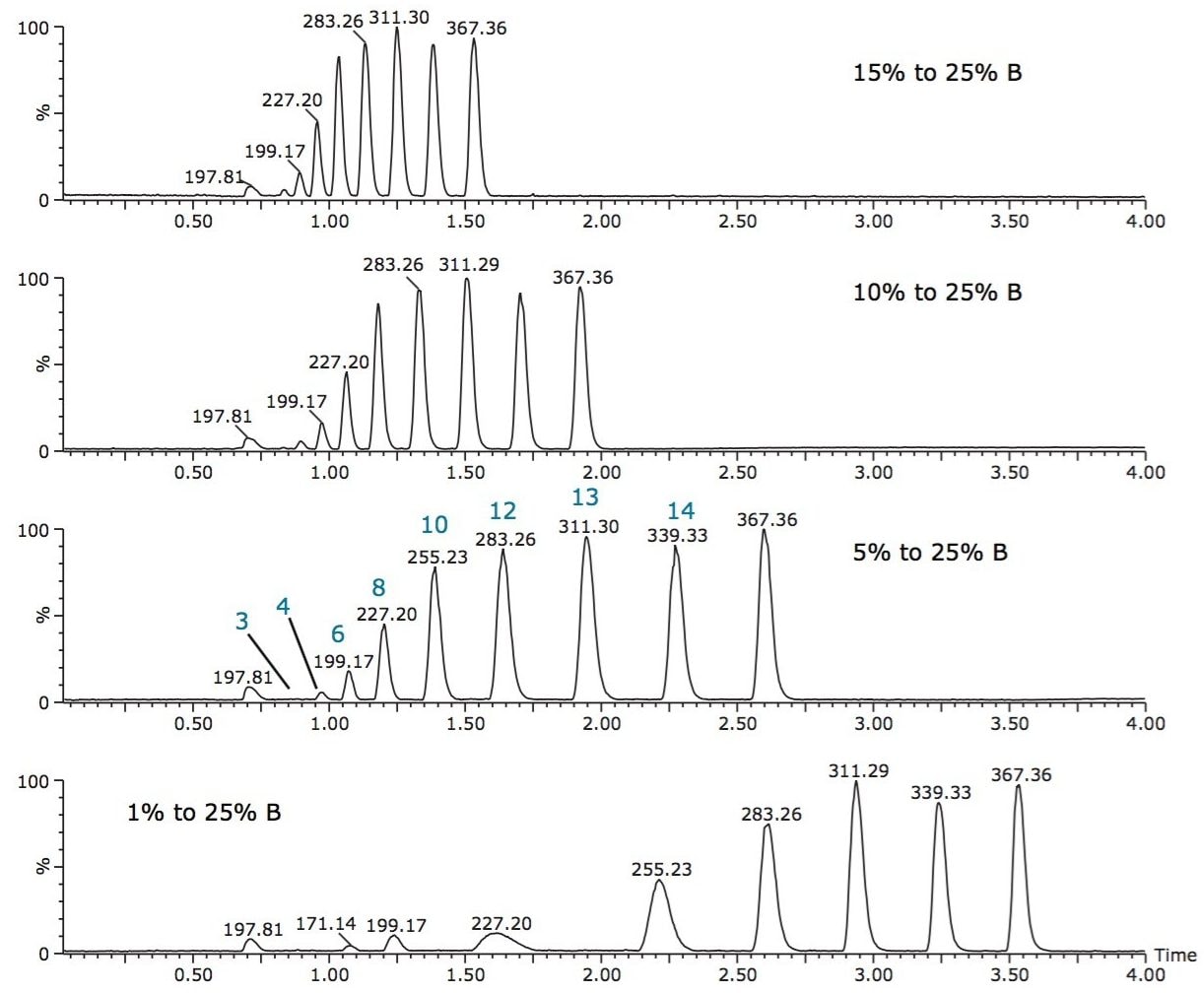
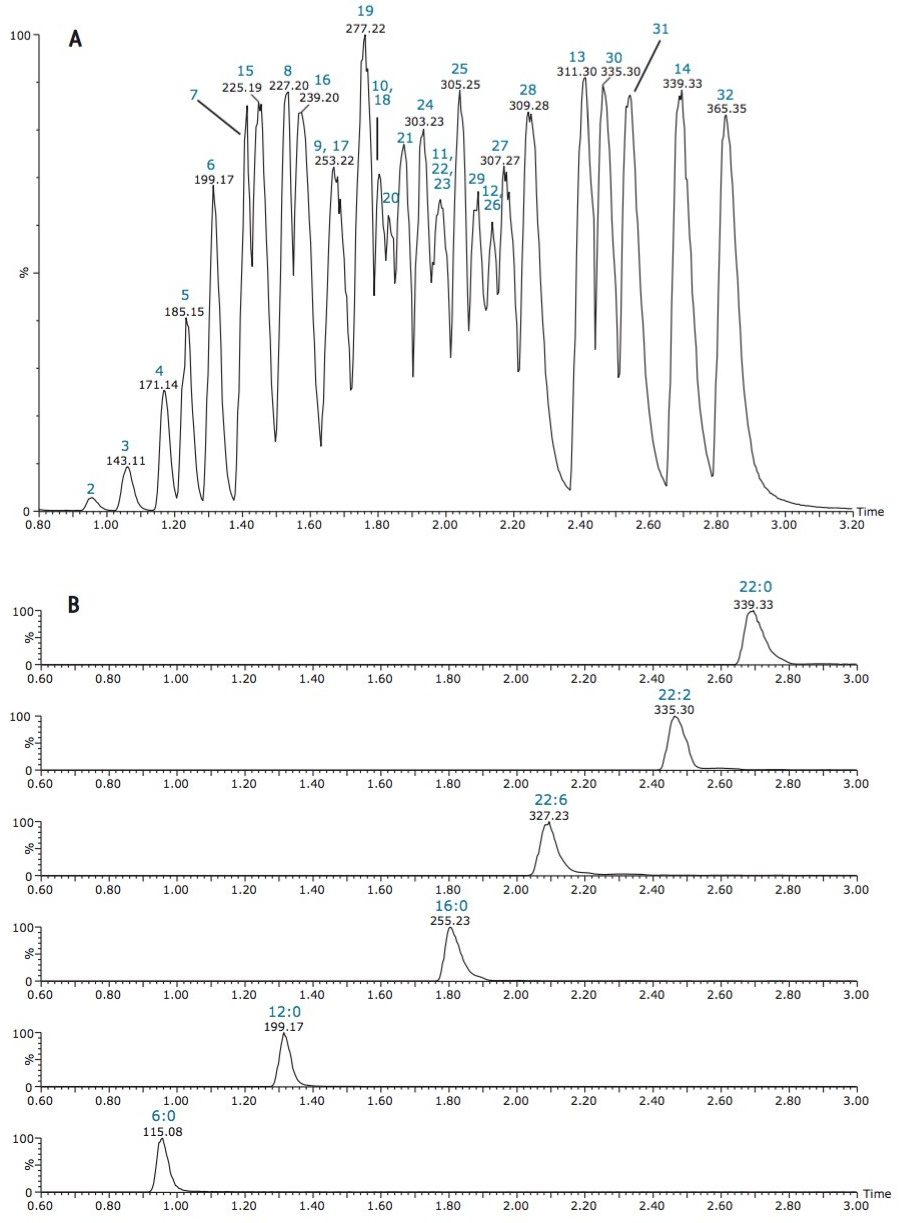
The isolation of free fatty acids (FFA) from biological materials is a complex task and precautions should be taken at all times to prevent or minimize the effects of hydrolyzing enzymes. After isolation, the typical chromatographic methods for analyzing fatty acids include gas chromatography/mass spectroscopy (GC-MS) and liquid chromatography-tandem mass spectrometry (LC-MS/MS). However, there are shortcomings associated with each of these methods.
For example, GC methods require derivatization of the fatty acids to hydrolyze and convert to methyl esters, which is time-consuming and risks re-arrangement of the fatty acids during derivatization, leaving doubt as to whether the esters formed are from FFA or intact complex lipids. Moreover, the GC-MS analysis of low volatile, very-long-chain fatty acids with high molecular weight (>C24) is a problem even after fatty acid methyl ester (FAME) derivatization.