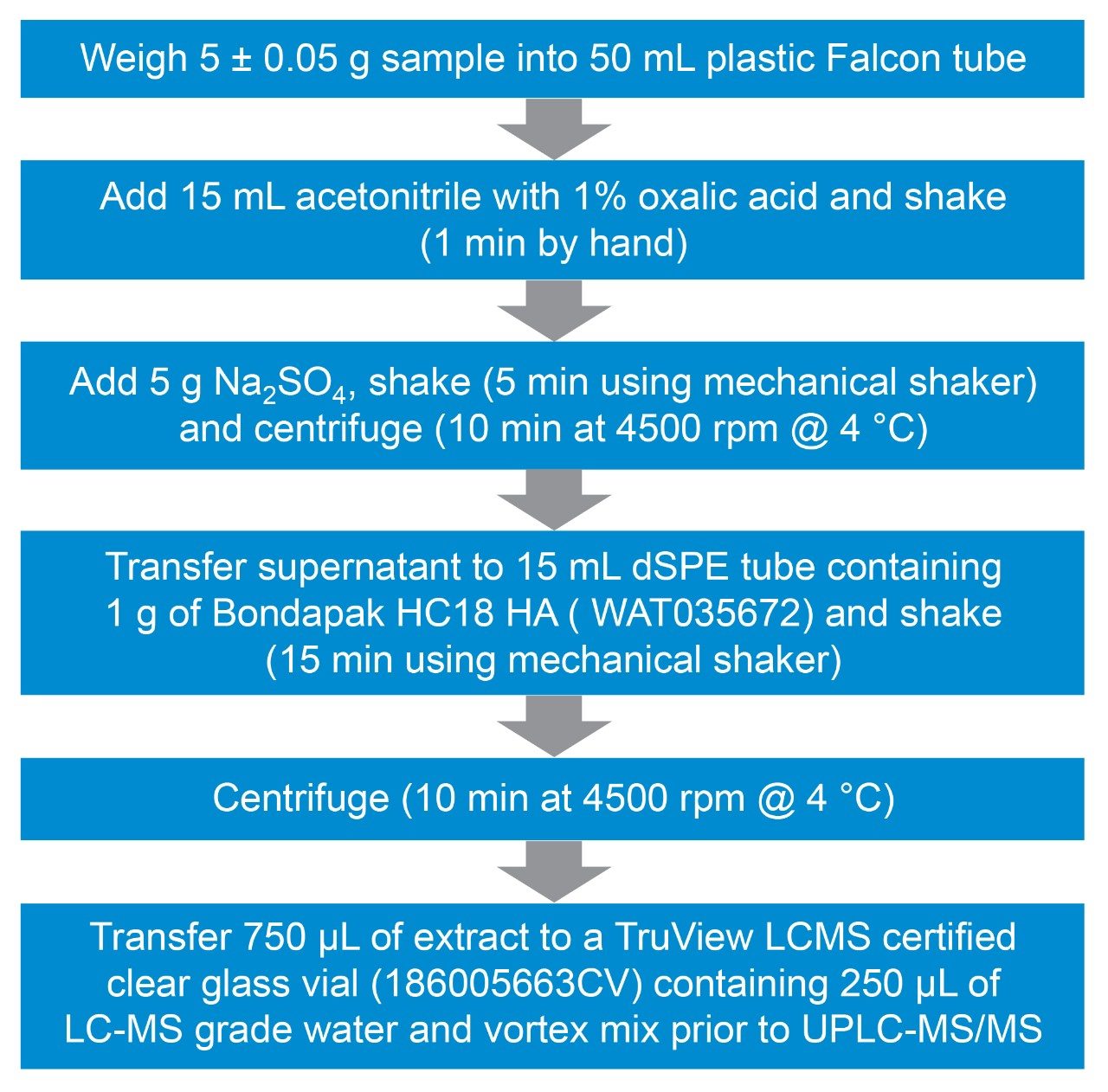
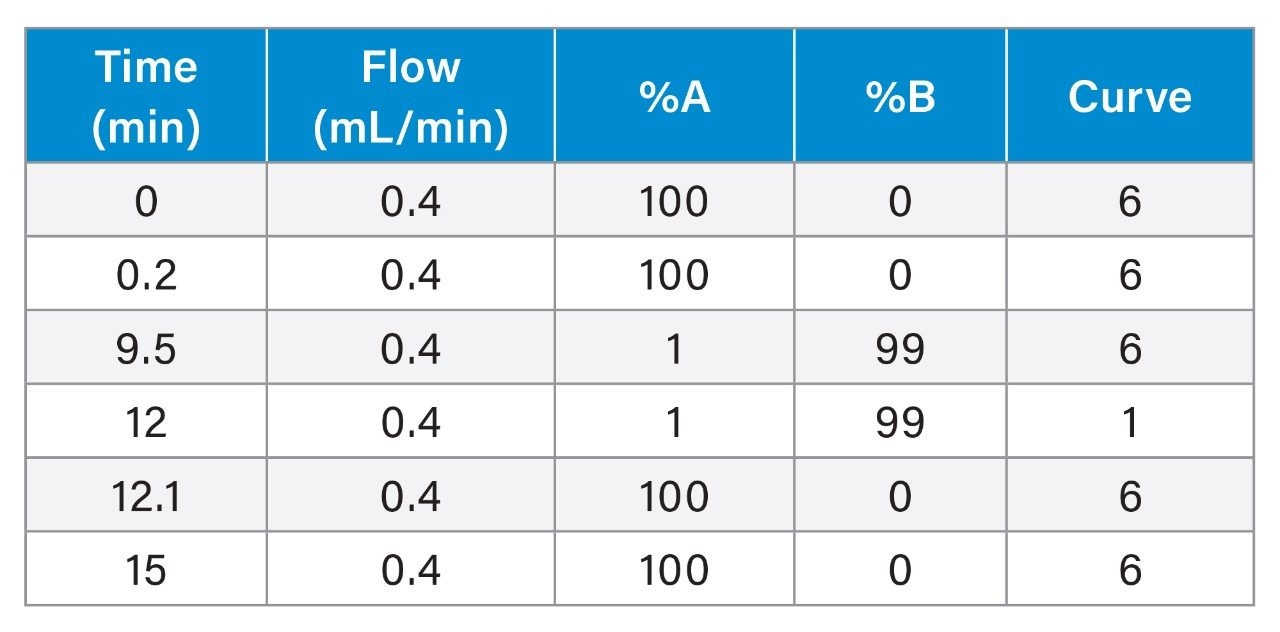
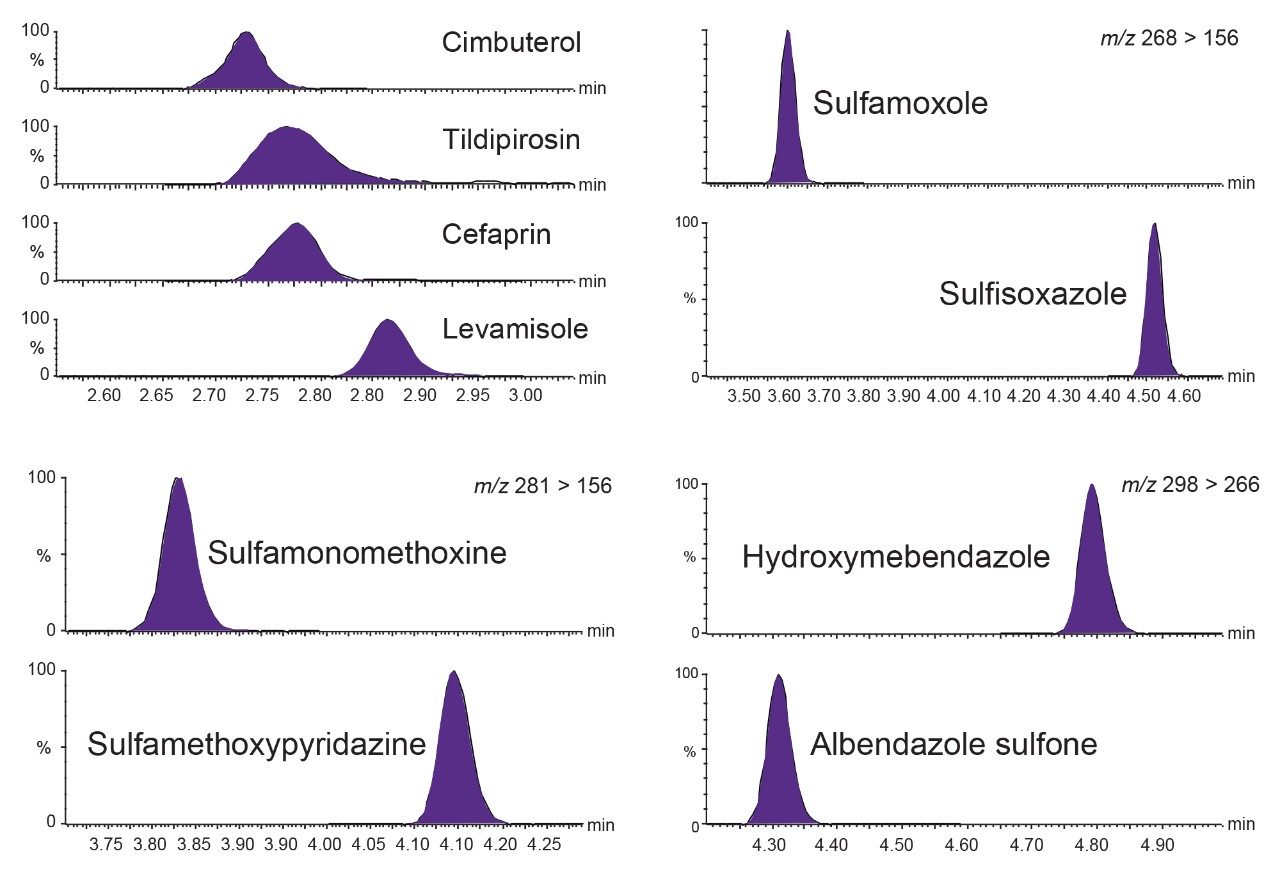
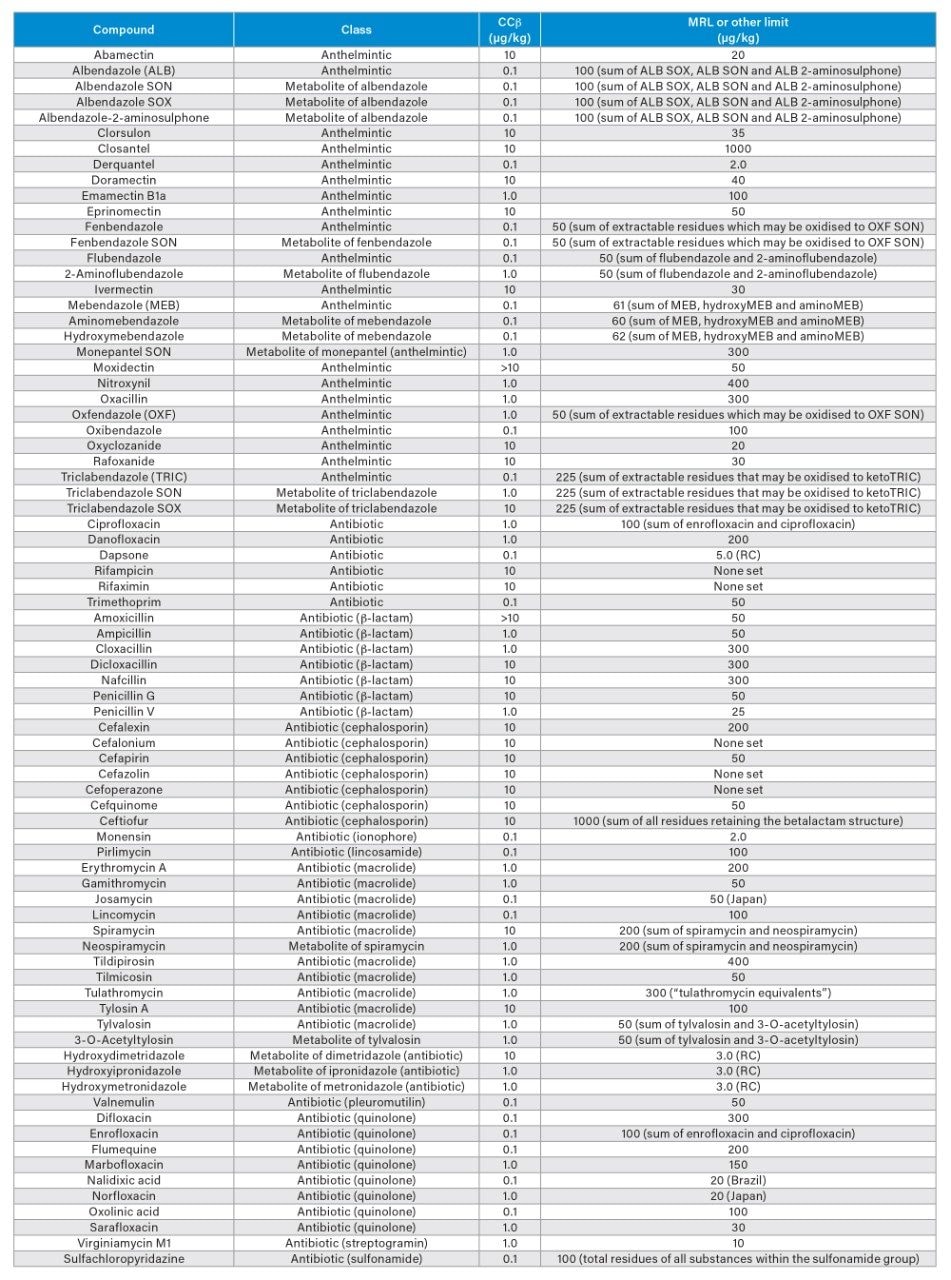
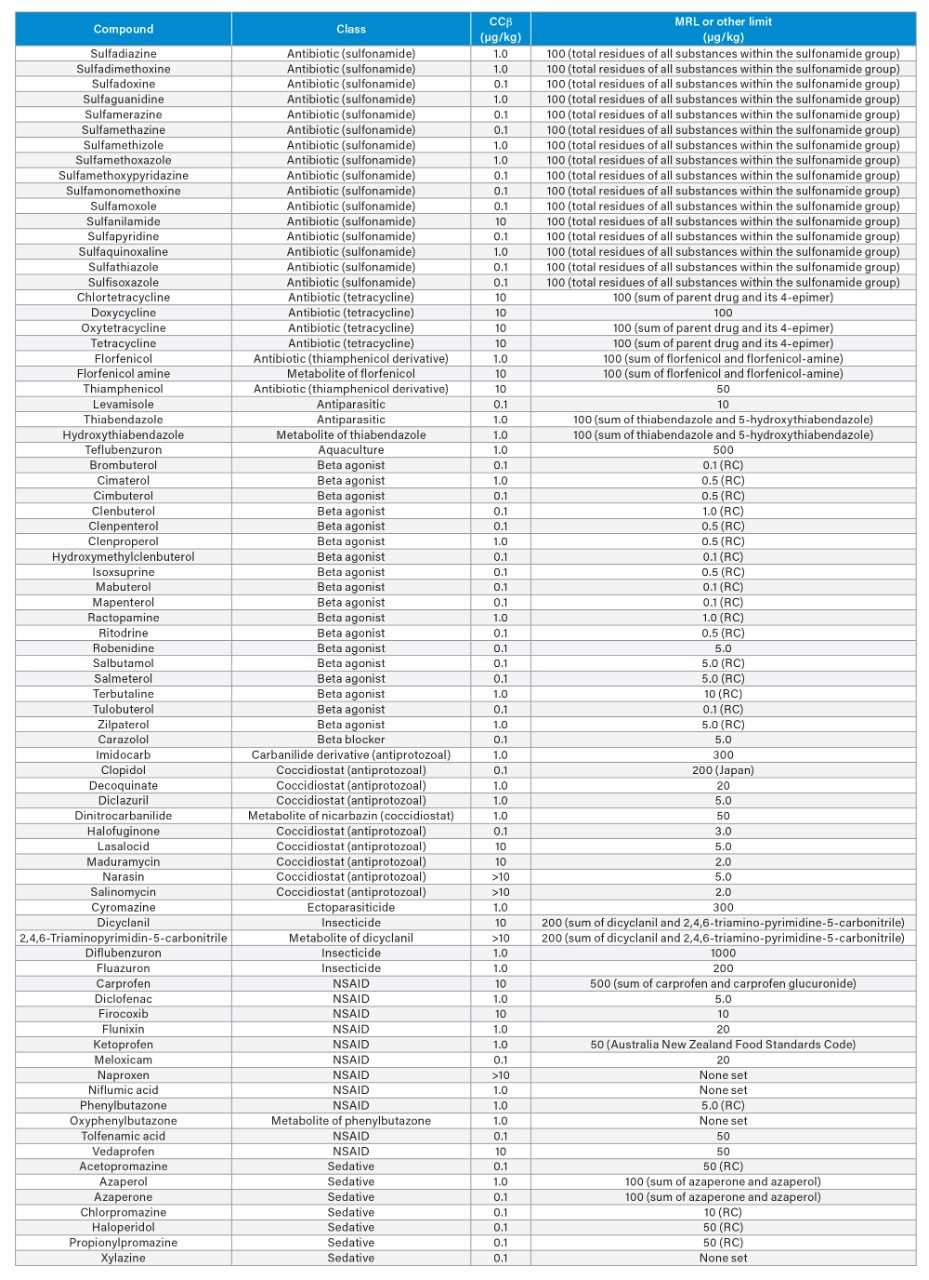
Validation Criteria
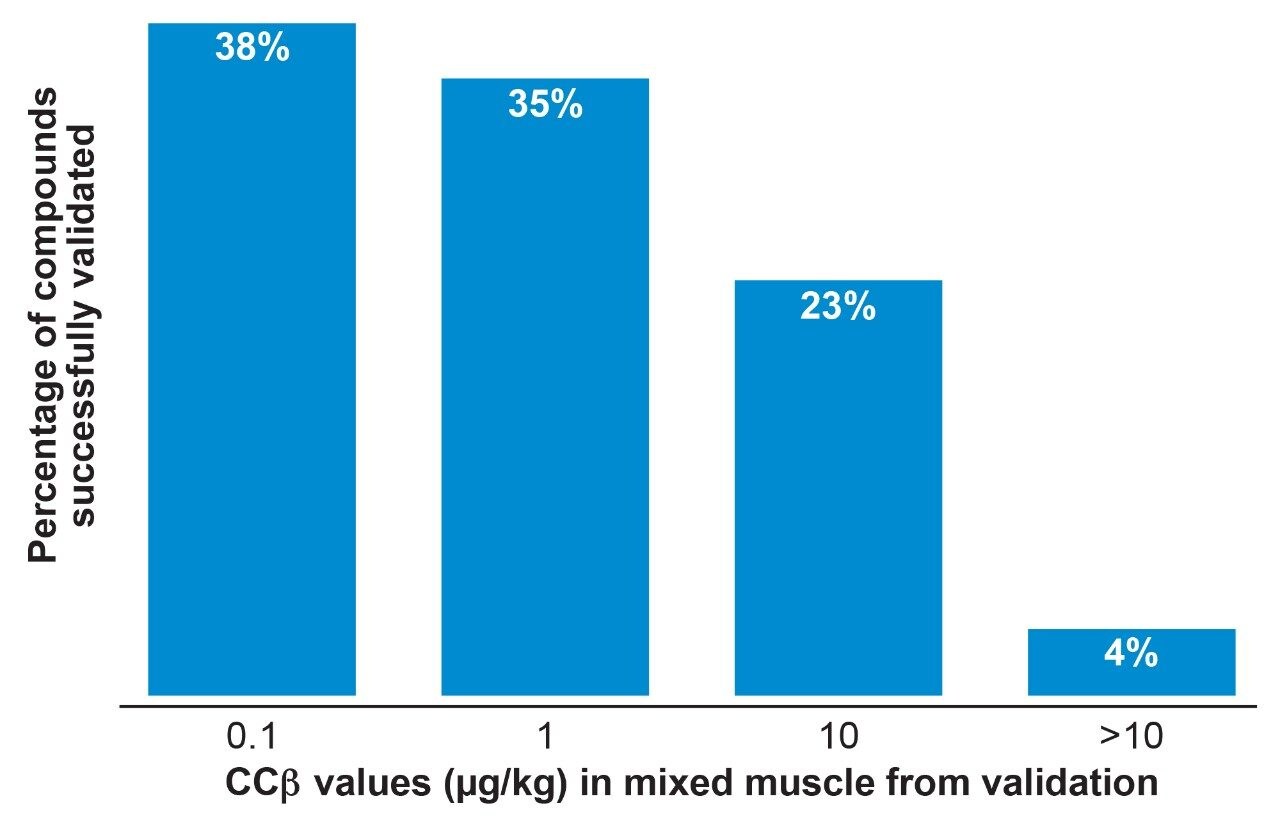
Detection capability (CCβ) is the smallest amount of a substance that may be detected in a sample with a false negative rate of 5%. For authorized pharmacologically active substances the CCβ shall be less than the MRL but for prohibited or unauthorized substances, the CCβ shall be as low as analytically achievable. The CCβ was evaluated by a comparison of the Threshold value (T) and Cut-off factor (Fm).
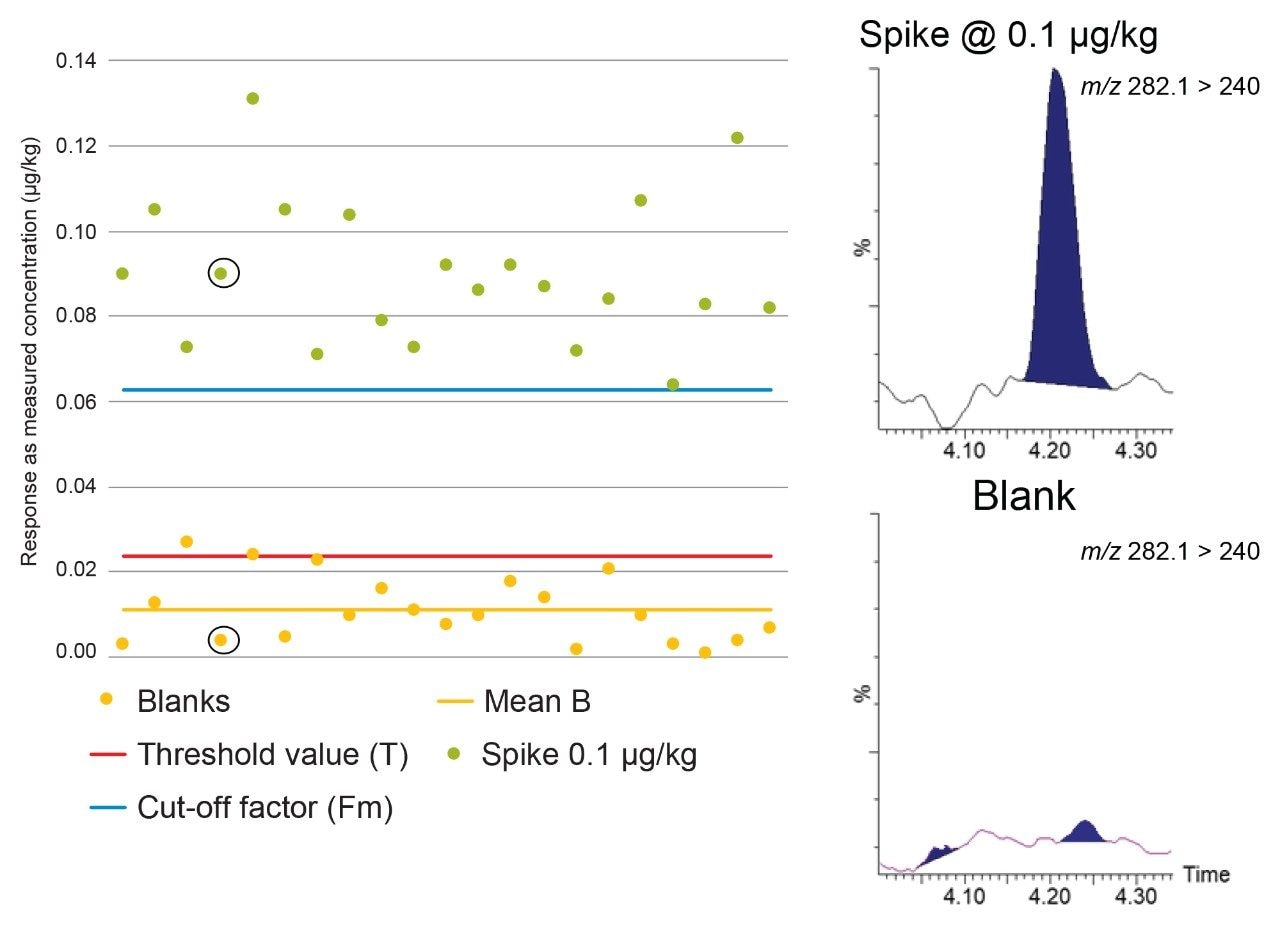
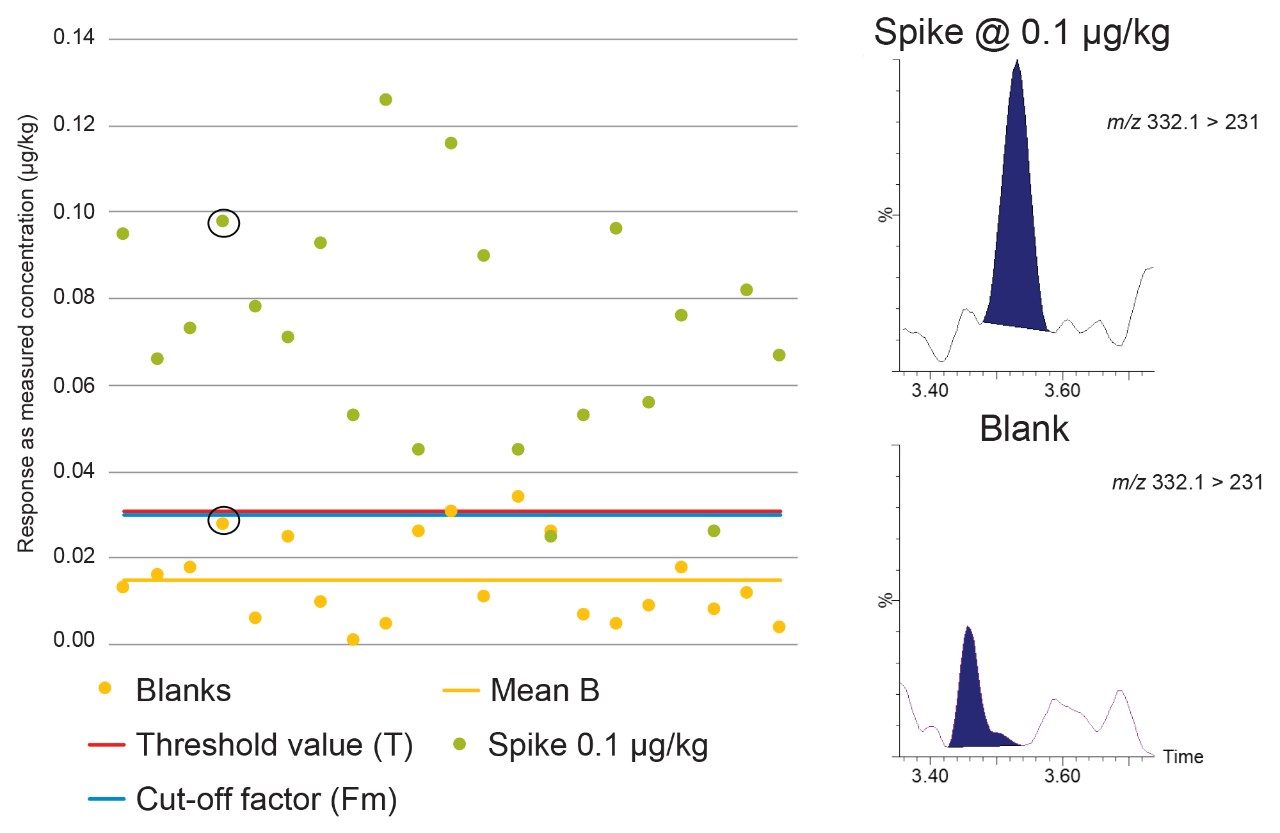
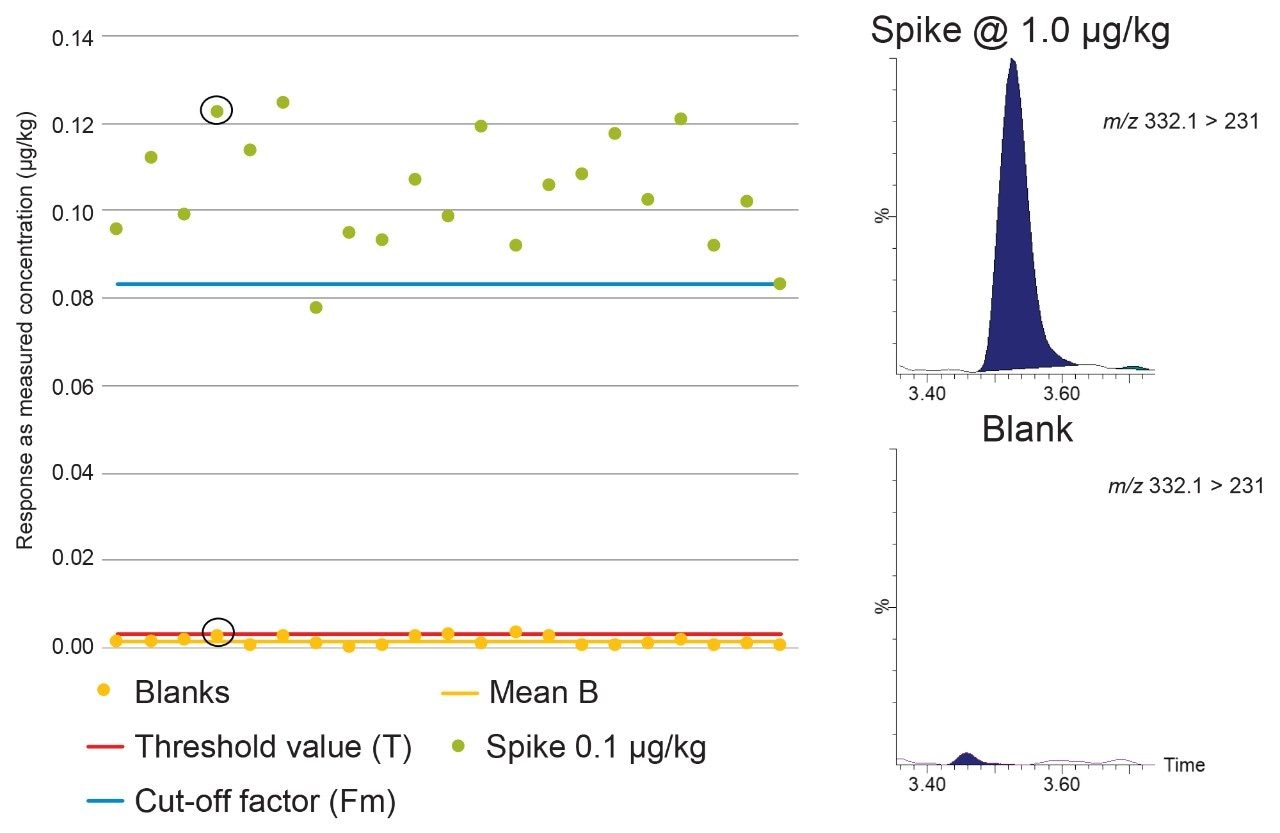
The Threshold value (T) is a value corresponding to the minimum analytical response above which the sample will be truly considered as positive. This parameter was determined by analyses of 21 blank samples from different origins and calculated using the equation:
T = B+(1.64×SDb),
which considers the mean value of the blank/noise “B” and the standard deviation of the blank/noise “SDb”. The 1.64 factor is the one-tailed Student t-value for infinite degrees of freedom at a significance level, β = 0.05. The Cut-off factor (Fm) was determined by analyses of 21 blank samples spiked at the level of interest for each analyte. The response was determined for each analyte and the Fm calculated using the following equation:
Fm = M–(1.64×SD),
which considers the mean response “M” and the standard deviation “SD” for each analyte.
According to Commission Decision 2002/657/EC, CCβ is validated when Fm>B. The rate of false positives is acceptable (i.e. 5%) when Fm>T. So, when B<T<Fm, the CCβ is truly below the spiked concentration, the STC being tested, indicating the false negative rate = 5% and the false positive rate <5%. When T>Fm, more than 5% of the spiked samples will be considered as negative so the CCβ cannot be established at that spiking concentration so the assessment is repeated at higher concentrations.
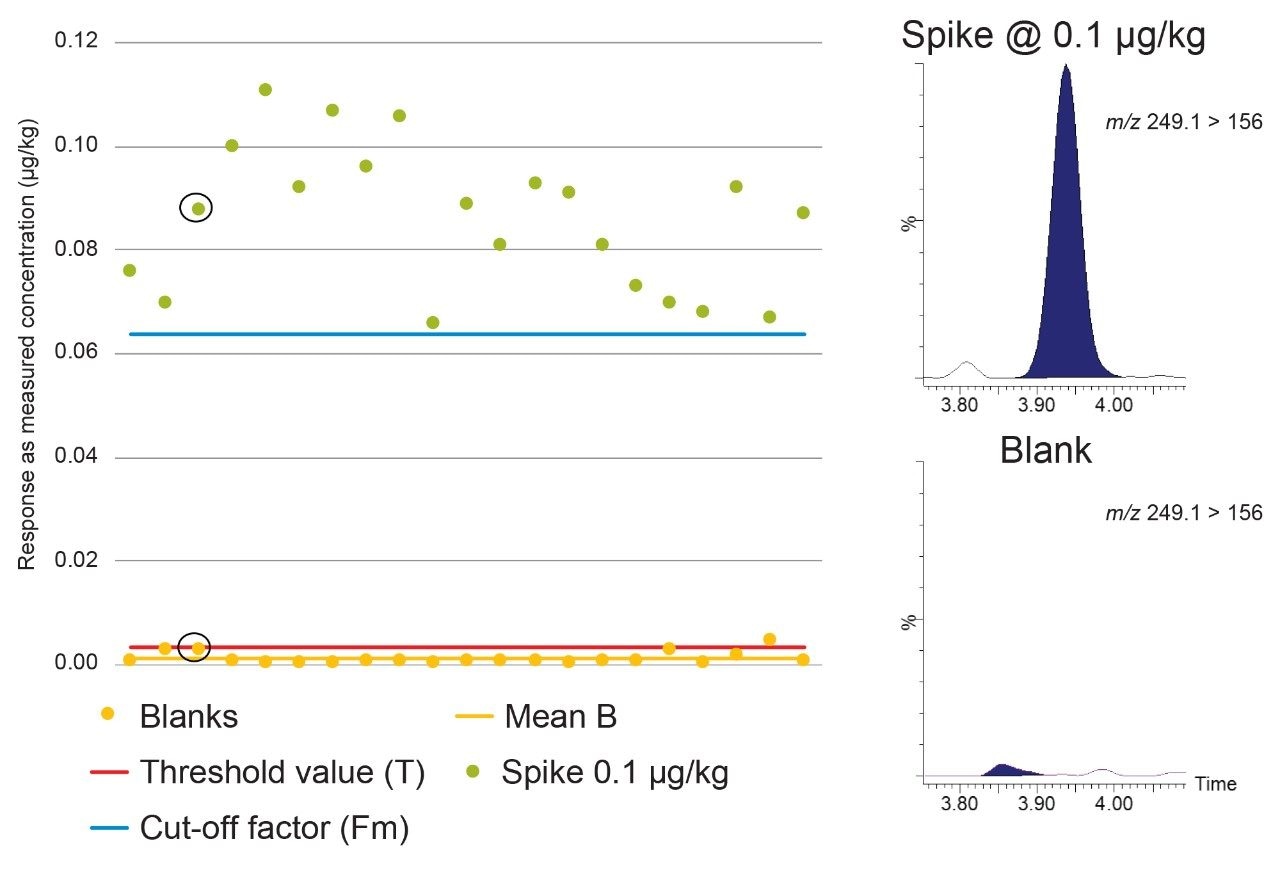
Figure 3 shows Blank (B), Threshold value (T), and Cut-off factor (Fm) for dapsone, a banned substance, in mixed muscle samples. Using the spikes at 0.1 µg/kg, B<T<Fm, so the STC can be established at 0.1 µg/kg, well below the RC of 5 µg/kg for muscle.