Most biotherapeutic proteins are produced by recombinant DNA technology using a highly qualified host cell system. Host cells express a large number of their own proteins that can easily contaminate the recombinant protein drug. Even after several orthogonal purifications steps, low levels (1–100 ppm) of host cell proteins may still remain in the final biopharmaceutical product. Since HCPs may induce an adverse immunogenic response, regulatory guidelines stipulate that HCPs must be identified and quantified to protect patient safety.
Characterization of HCPs in protein drugs is required before a biopharmaceutical can be accepted by the regulatory agencies worldwide. For example, in 2008, the European Medicines Agency (EMA) approved a recombinant form of human somatropin only after the manufacturer added additional purification steps to remove the HCPs responsible for immunogenic response in patients.1 The same agency rejected an interferon biosimilar in 2006 because of insufficient validation for immunogenicity testing.
Since HCPs are trace contaminants in the biopharmaceutical product, any analytical methods used to measure them must be effective over a wide dynamic range, typically from 4 to 6 orders of magnitude. Some widely used analytical methods, such as process-specific ELISAs and Western blots,2 require prior knowledge of the composition of HCP contaminants. In addition, process-specific immunoassays are both time consuming (e.g., 6 months) and expensive to develop (> $100K). Further, immunochemical assays cannot readily adapt to evaluate biopharmaceutical products from different cell types and purification protocols.
Two-dimensional gel electrophoresis coupled to fluorescent staining,3,4 another popular method for HCP analysis, is semi-quantitative, has limited dynamic range (2 to 3 orders of magnitude), and requires mass spectrometry or another off-line technique for HCP identification. Although commercial ELISA kits are readily available, they are developed for generic HCP monitoring, are less specific than the process-specific immunoassays, and cannot offer complete coverage for all the existing HCPs present in the sample.5,6
An organization that demonstrates the ability to accurately identify and monitor HCPs in biotherapeutic samples is more likely to meet the regulatory requirements and to produce better quality products.
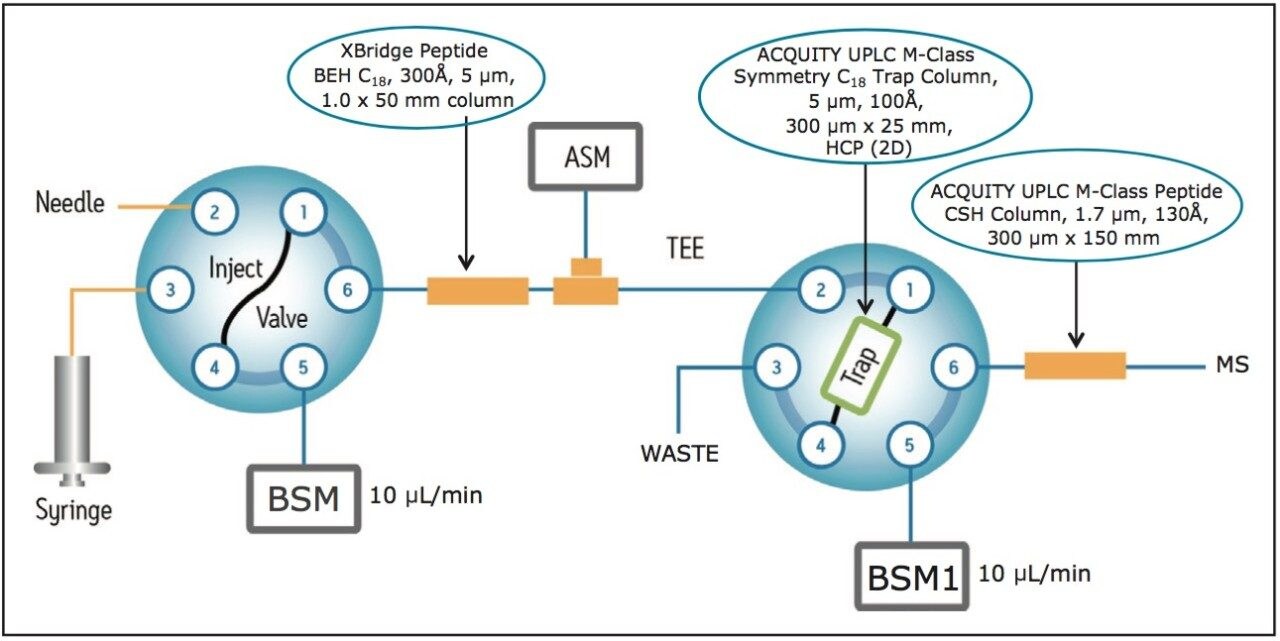
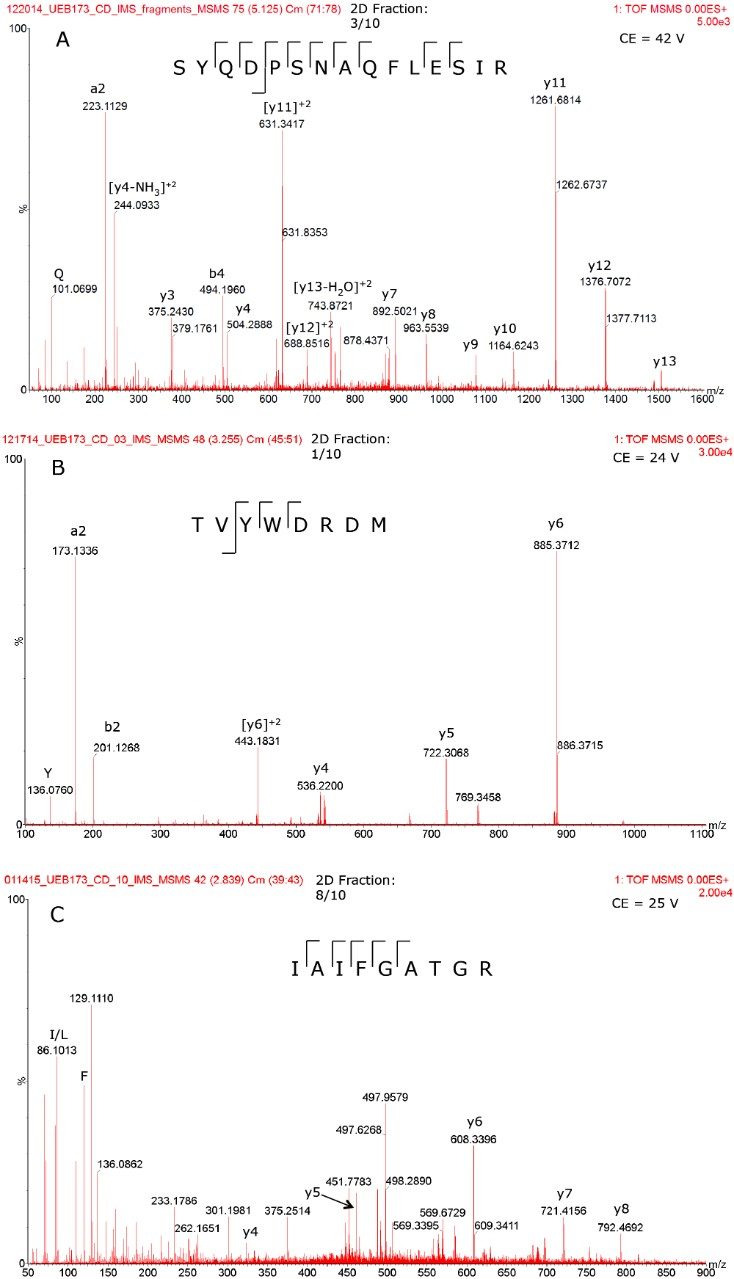
Here, a generic LC-MS assay that offers comprehensive HCP identification and quantification for biotherapeutic protein samples is described. The assay uses a bottom-up peptide-based strategy for HCP identification. The biopharmaceutical product is digested into peptides and analyzed with an on-line two-dimensional LC system coupled to a high-resolution, high-mass-accuracy ion mobility enabled mass spectrometer for protein identification and quantification.
For a successful LC-MS assay for HCP analysis, multiple dimensions of separation are clearly beneficial as a single analytical technique does not offer the required dynamic range. In contrast to traditional 2D-chromatography schemes based on strong cation exchange (SCX) and low-pH reversed-phase7 separations, the 2D-LC configuration used here couples a high-pH (pH 10) reversed-phase (RP) separation to a low-pH (pH 2.5) RP separation.8-13
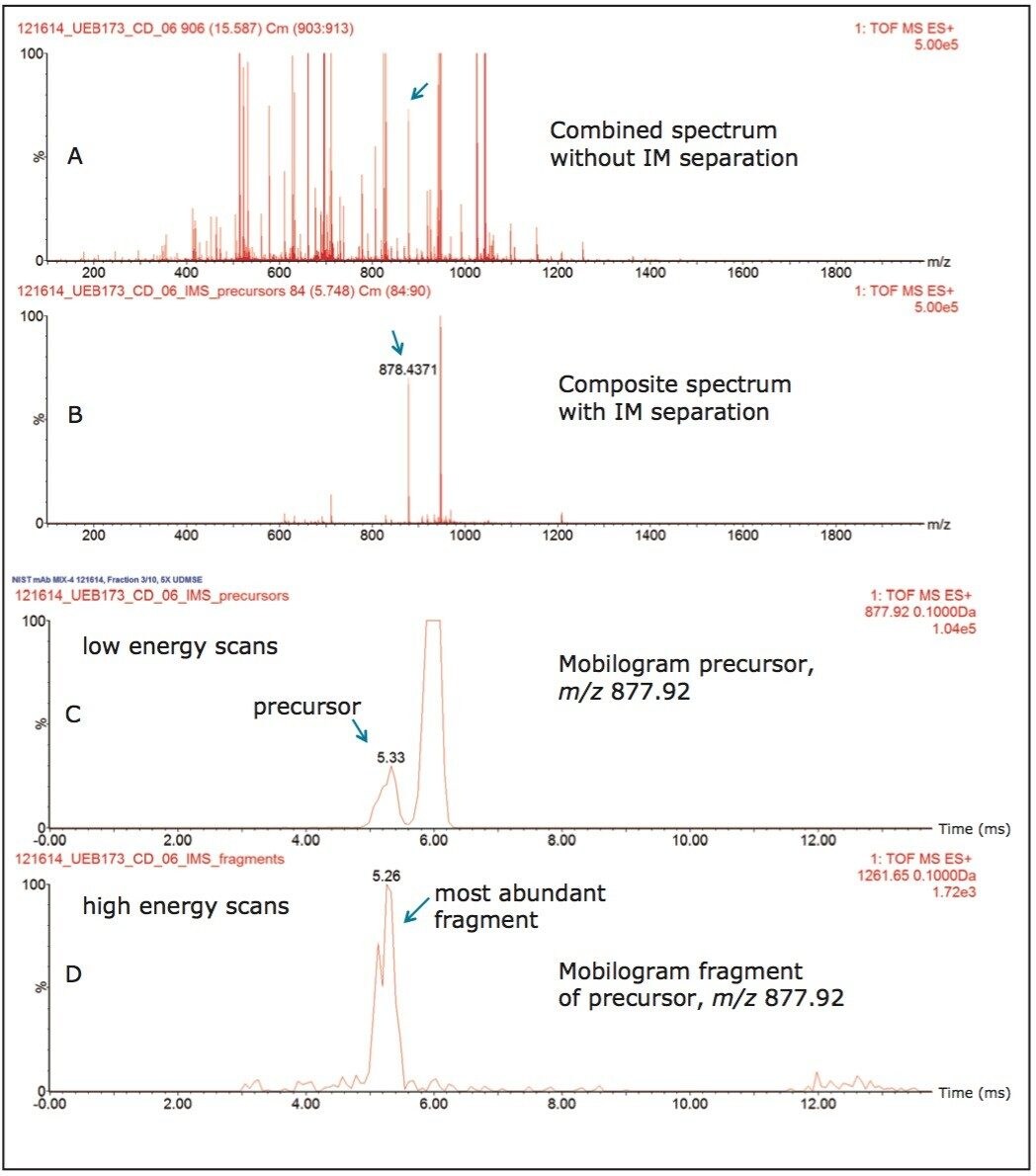
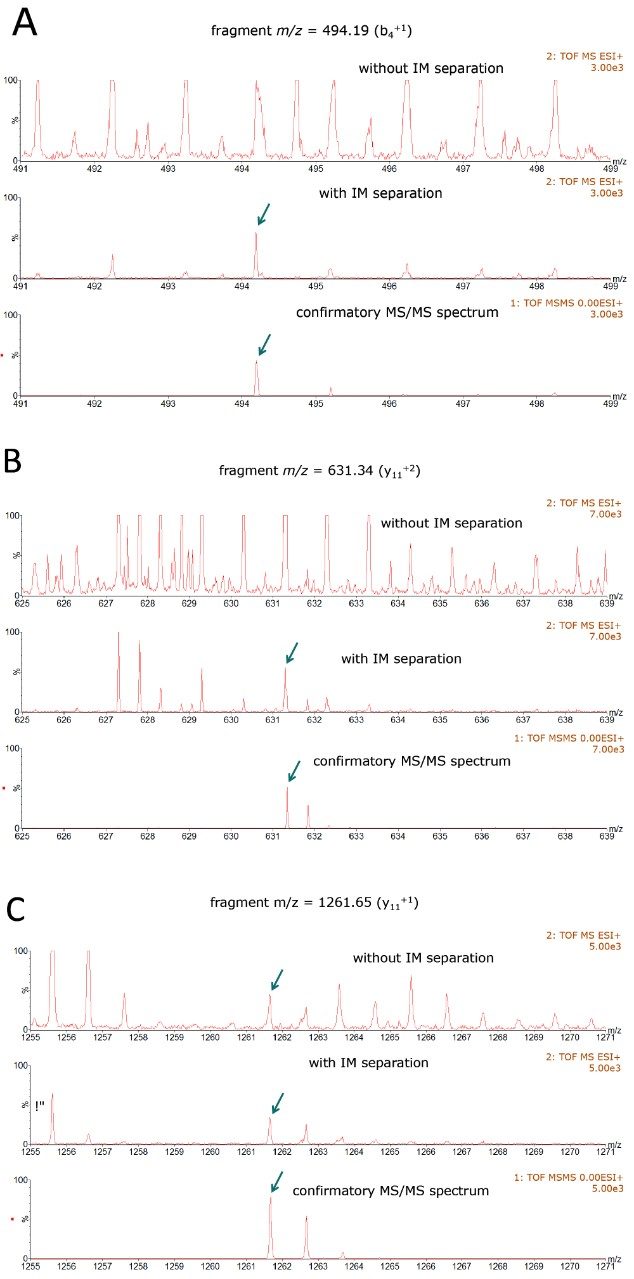
This 2D-RP/RP system achieves high-peak-capacity chromatographic separations, and thus is able to resolve the high complexity typically encountered in HCP analysis. In addition, a multiplexed data acquisition method using travelling wave (T-Wave) ion mobility separation of peptide precursor ions, followed by their fragmentation using ion mobility-specific collision energies,15 is utilized in the mass spectrometric data acquisition, such that low abundance HCP peptides can be reproducibly sampled and identified.
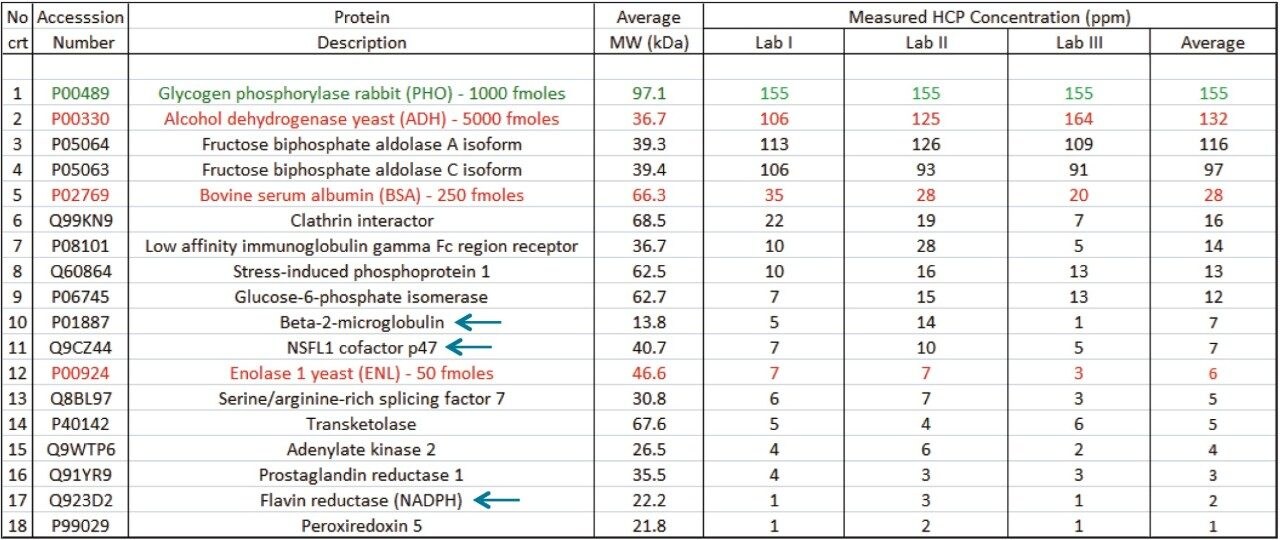
The ability of this 2D UPLC-MS system to identify and measure low-abundance HCPs (1 to 100 ppm) is demonstrated by analyzing a highly-purified monoclonal antibody that has been made available from the National Institute of Standards and Technology (NIST).