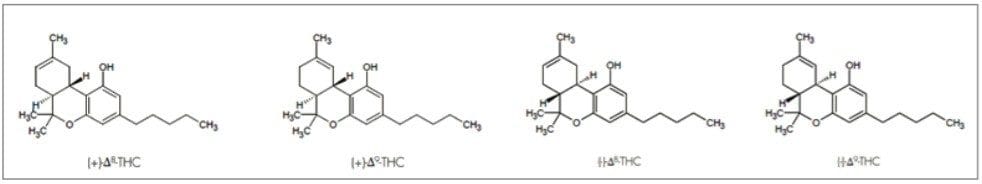
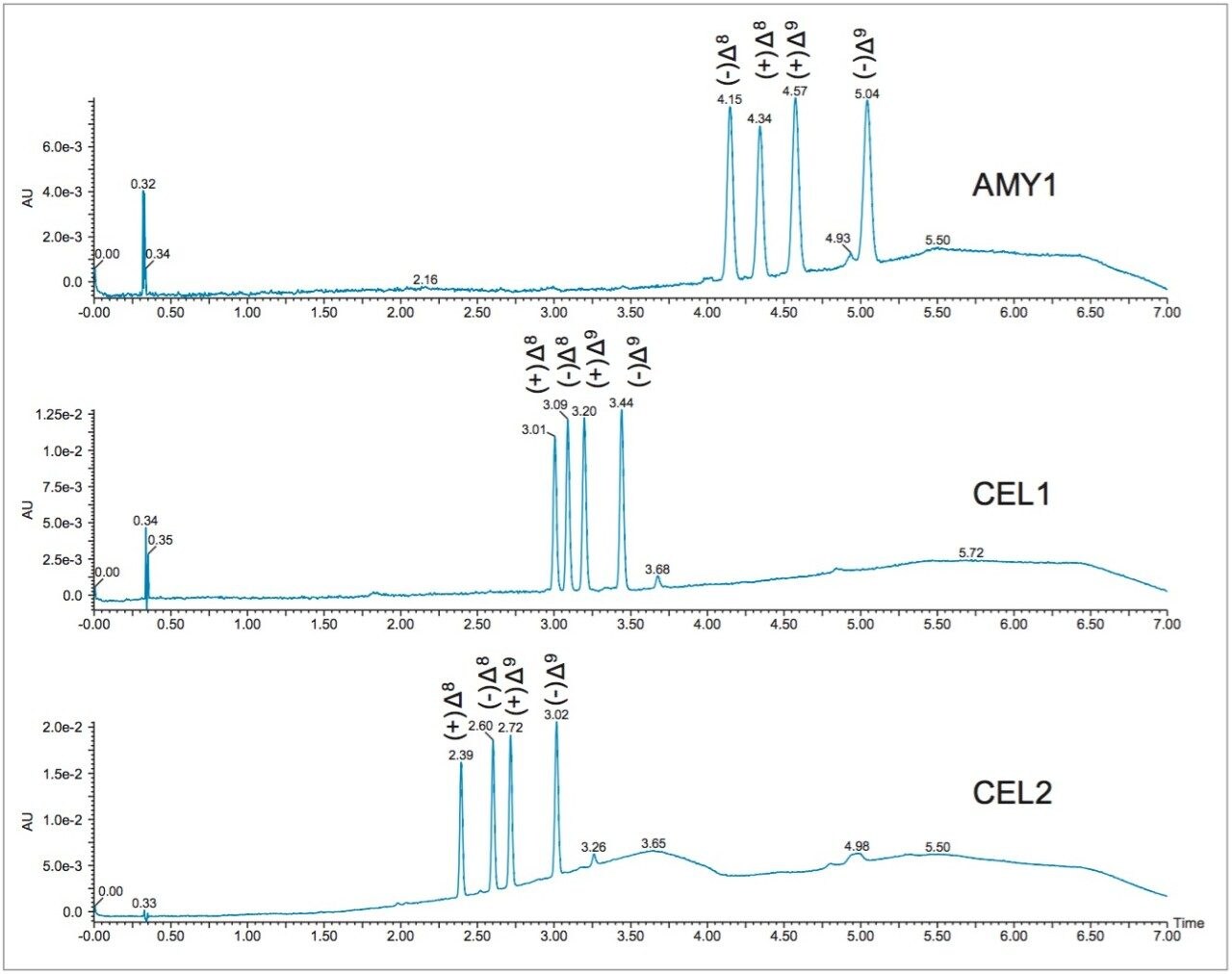
Optimization of the mobile phase conditions usually involves either focusing the gradient or determining isocratic conditions. Isocratic methods are ideal because they are easy to develop and no equilibration is required between runs, improving productivity. Using retention times and gradient slope, and compensating for system and column volume delay, the co-solvent percentage at elution was determined for the separation on the AMY1 column.
With a gradient delay of 0.34 min, gradient slope of 3.6%/min, and 2% starting percentage, the co-solvent percentage
at elution of the first peak at 4.15 was calculated using the following equation:
%Co-solvent at elution = (retention time – gradient delay) x gradient slope + starting %
%Co-solvent at elution = (4.15 – 0.34 min ) x 3.6%/min + 2%
% Co-solvent at elution = 15.7%
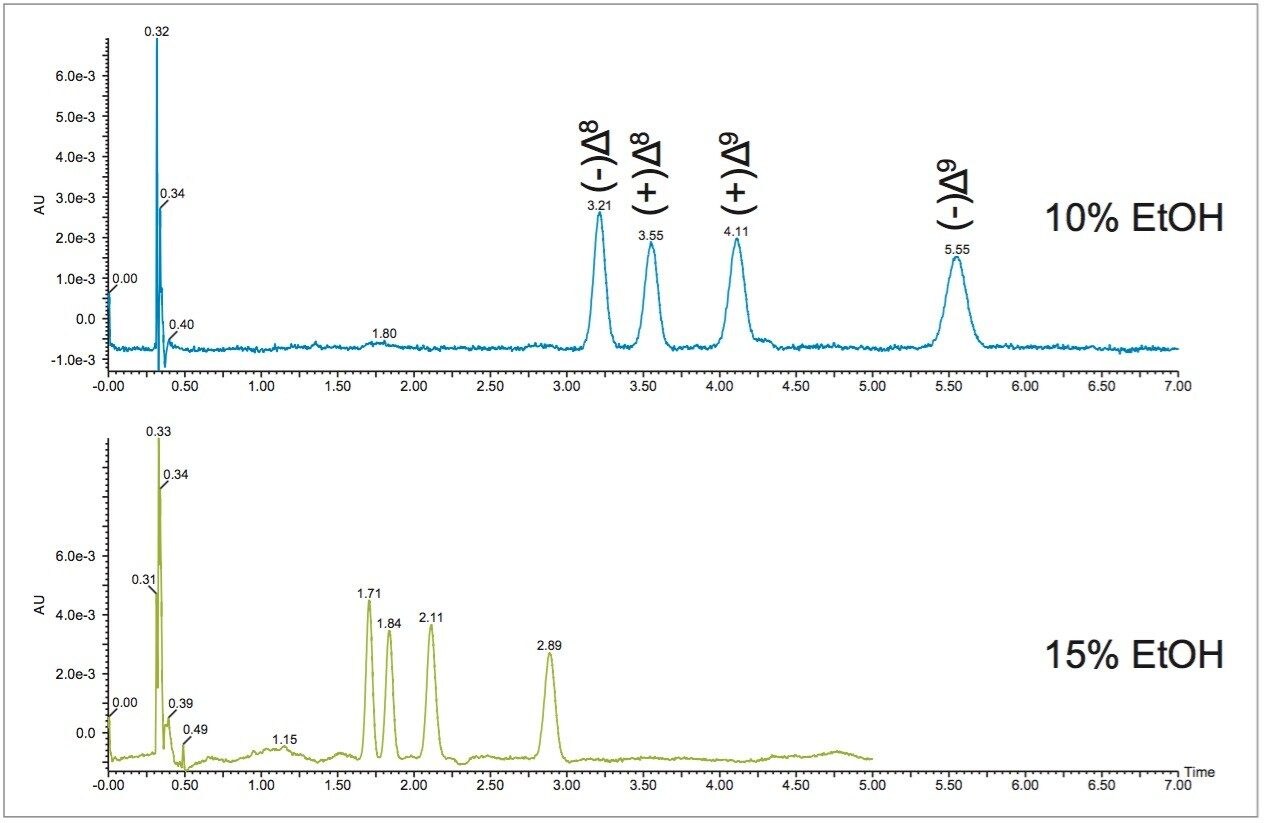
In SFC, usually the best starting point for optimization is 5% below the calculated percentage. Therefore, the initial isocratic method was run at 10% ethanol. Figure 3 shows separation of the THC isomers at 10% and 15% ethanol conditions. At 15%, the separation of the positional and stereo isomers of THC is achievable in less than 3 minutes. Comparatively, the liquid chromatography separation of these compounds does not result in baseline resolution of all four enantiomers, even with a relatively long 23 minute run time.3