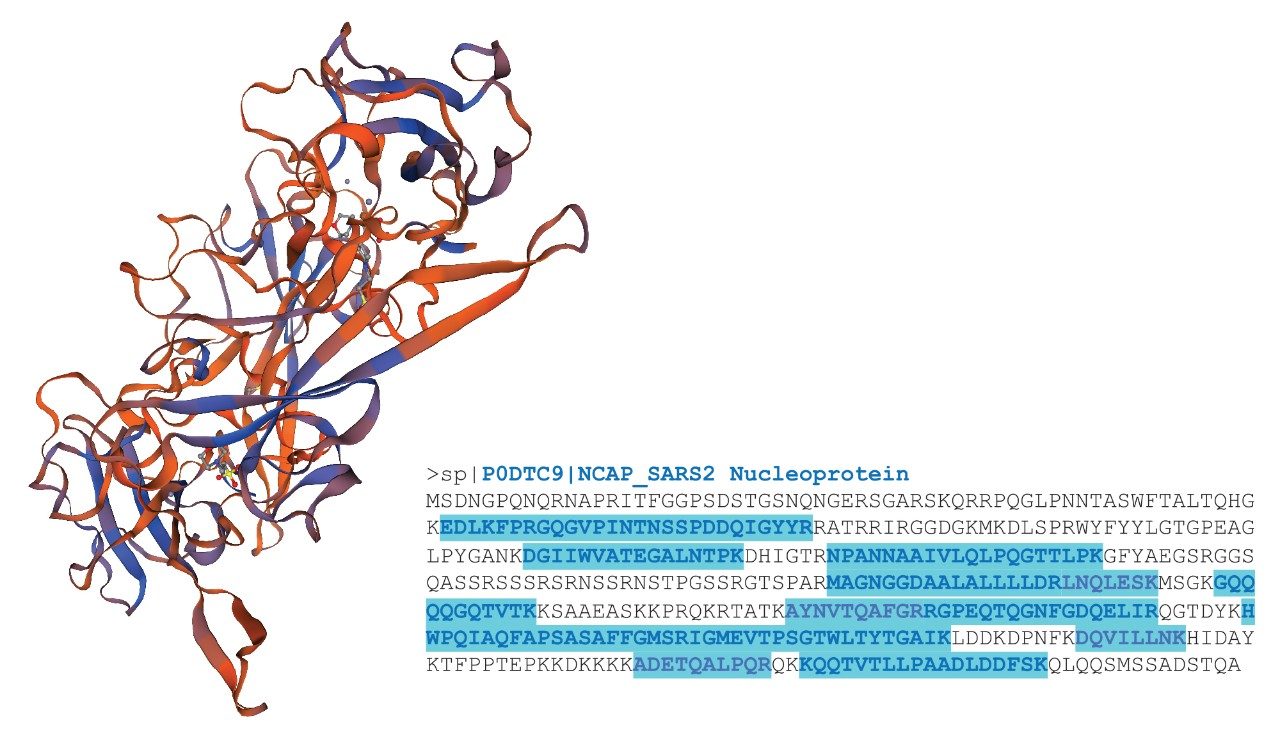
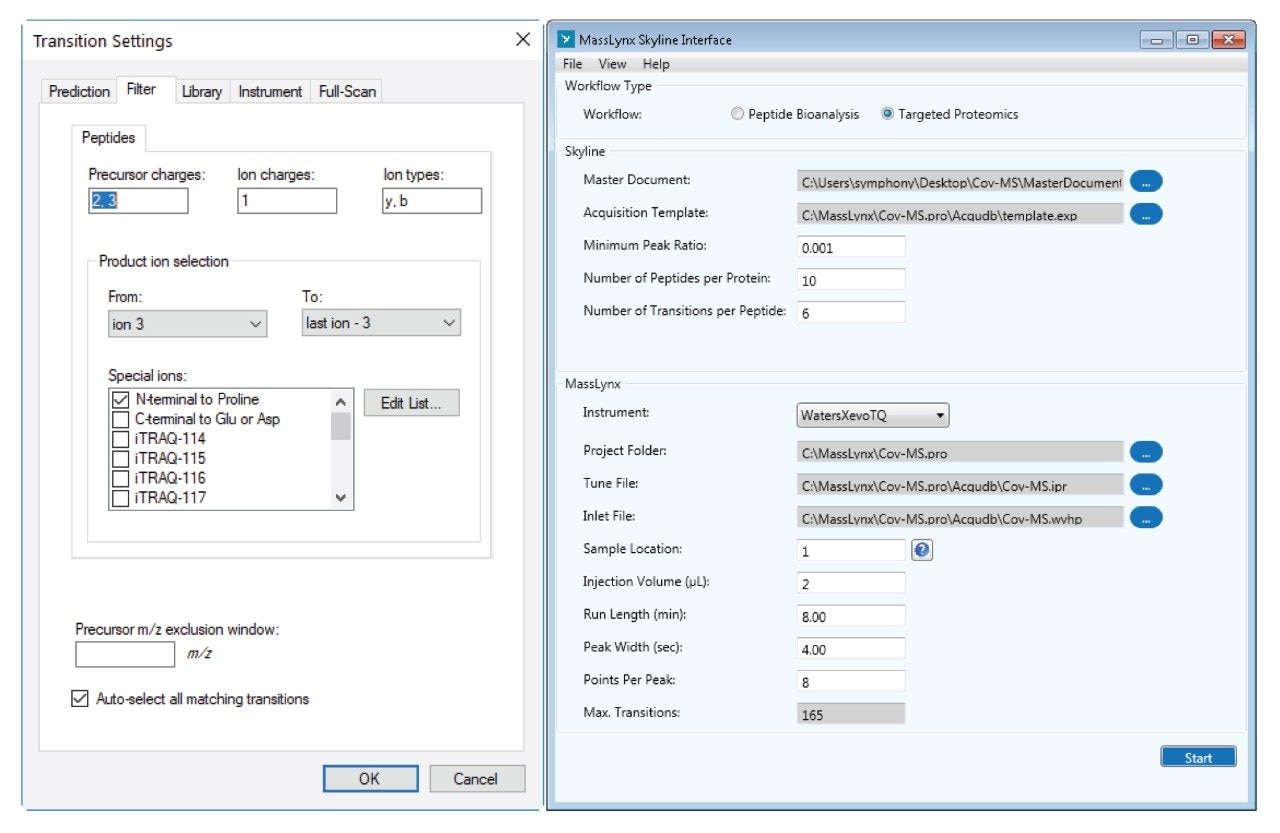
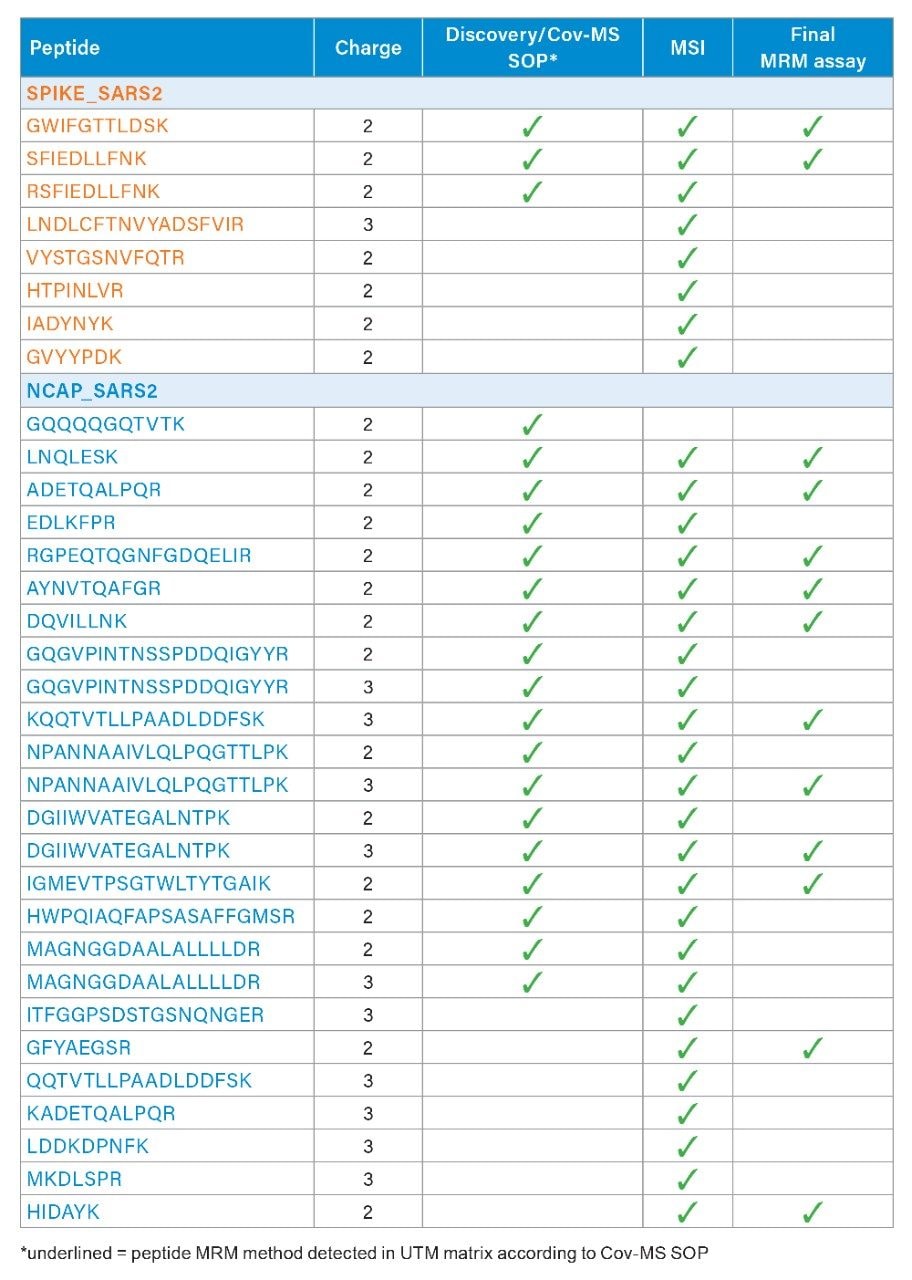
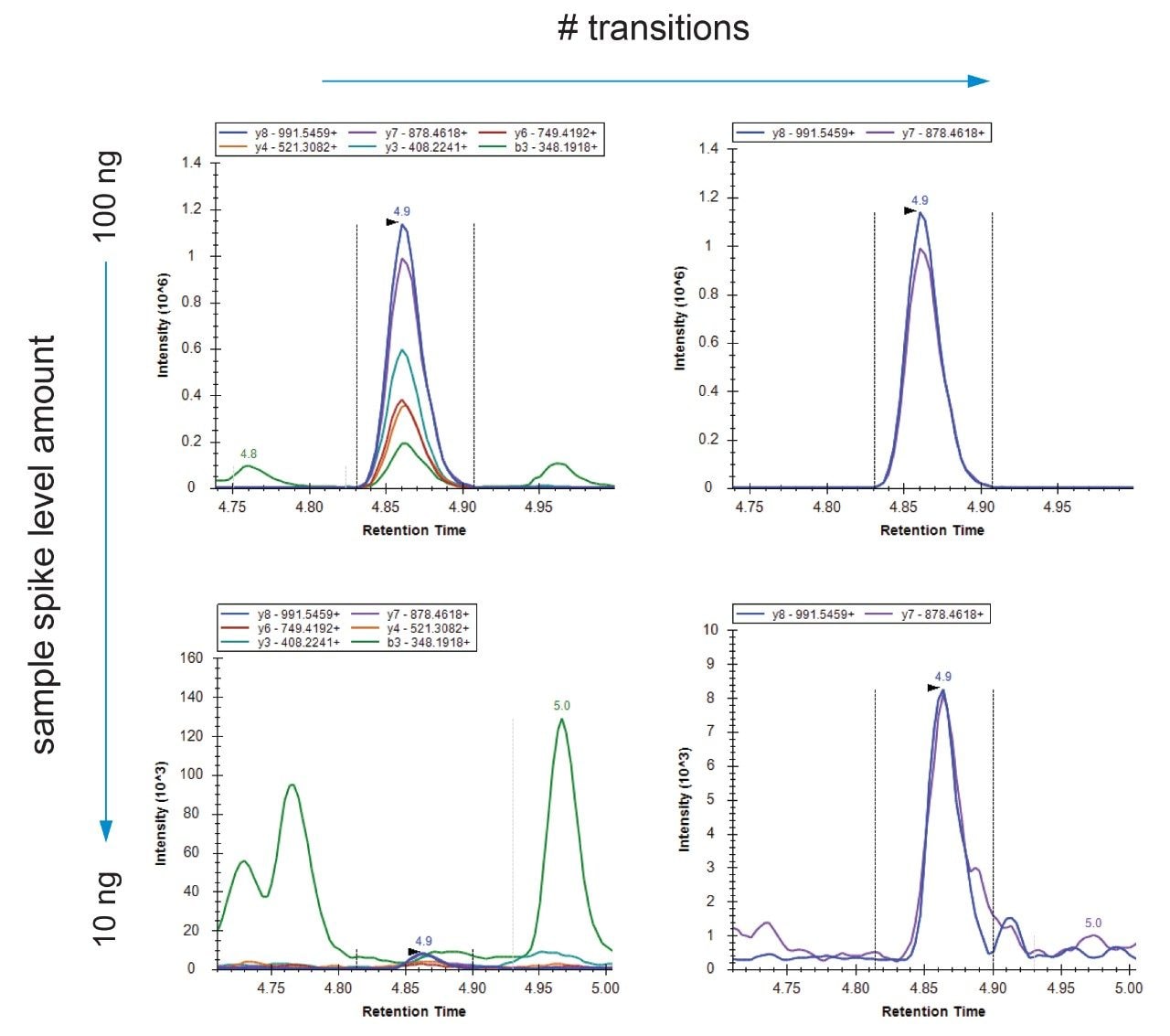
The NCAP amino acid sequence and coverage, as defined in the original Cov-MS standard operation procedure (SOP), is shown in Figure 1. Together with the primary amino acid sequence of the SPIKE protein, these form the basis for the MRM selection and optimization process. The MassLynx Skyline Interface (MSI) process was followed for automated optimization and fine tuning of a tandem quadrupole MRM method,7 as well as the detection of any additional candidate signature peptides. In short, MSI conducts an automated four-step process whereby first the retention time of the peptides is determined. Next, the most sensitive precursor/product ion pairs (MRM transitions) for every peptide are determined using a default calculated collision induced dissociation (CID) fragmentation energy. This is followed by optimization of the individual transition collision energies. Finally, an MRM method with appropriate acquisition windows is created. As shown on the left hand side of Figure 2, the appropriate peptide and transition settings are specified within a Skyline document. Next, as shown on the right hand side of Figure 2, the document is specified in MSI alongside with other methods files (tune page, acquisition method, LC gradient), sample position and injection volume.