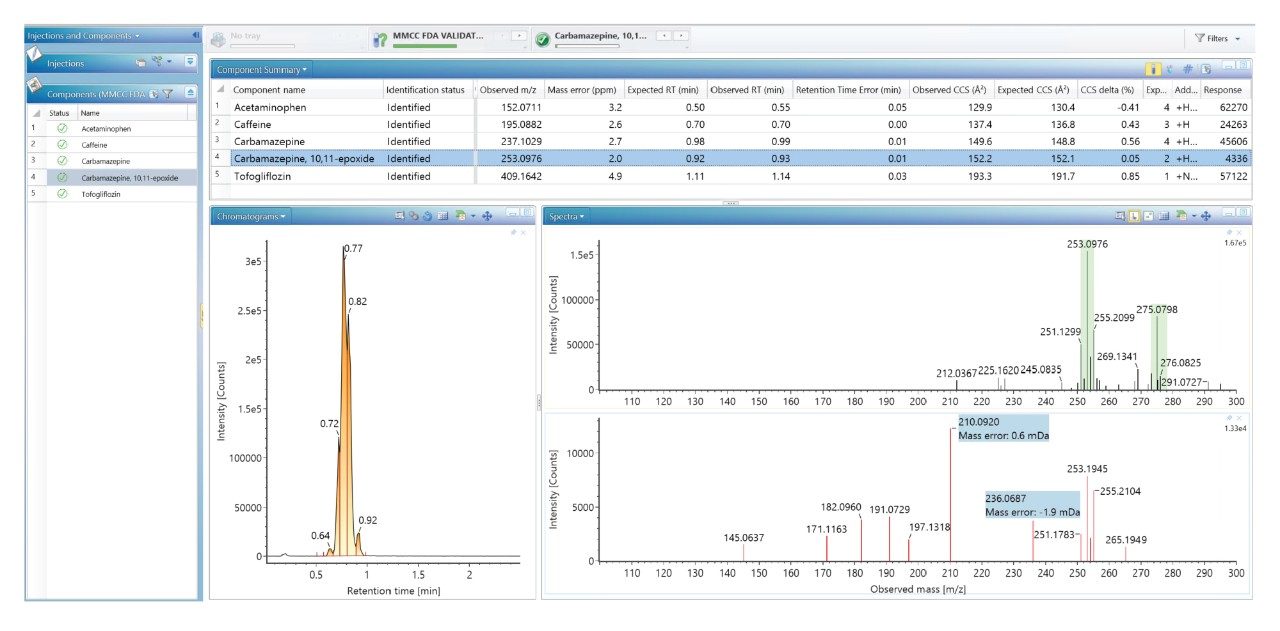
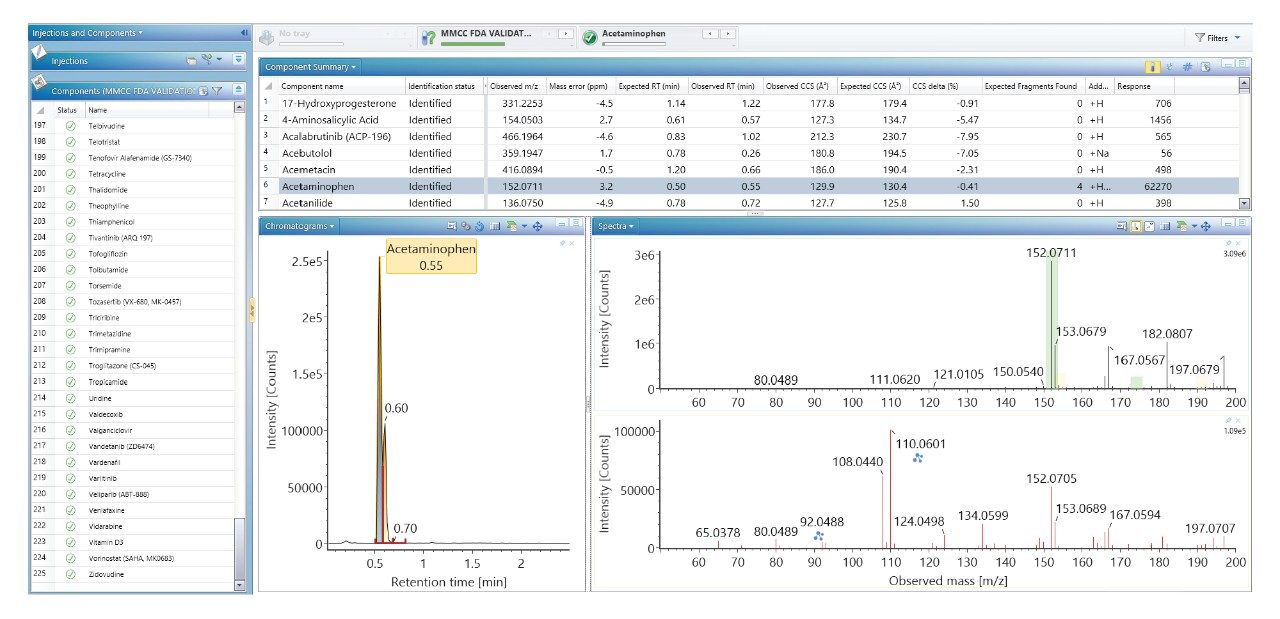
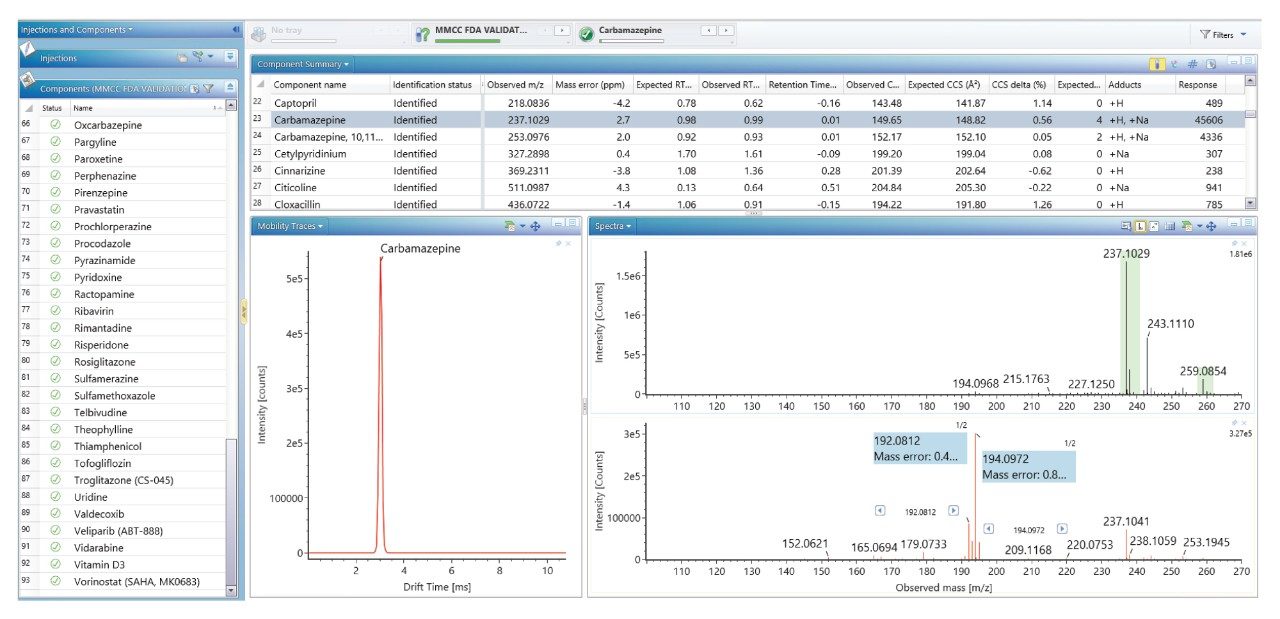
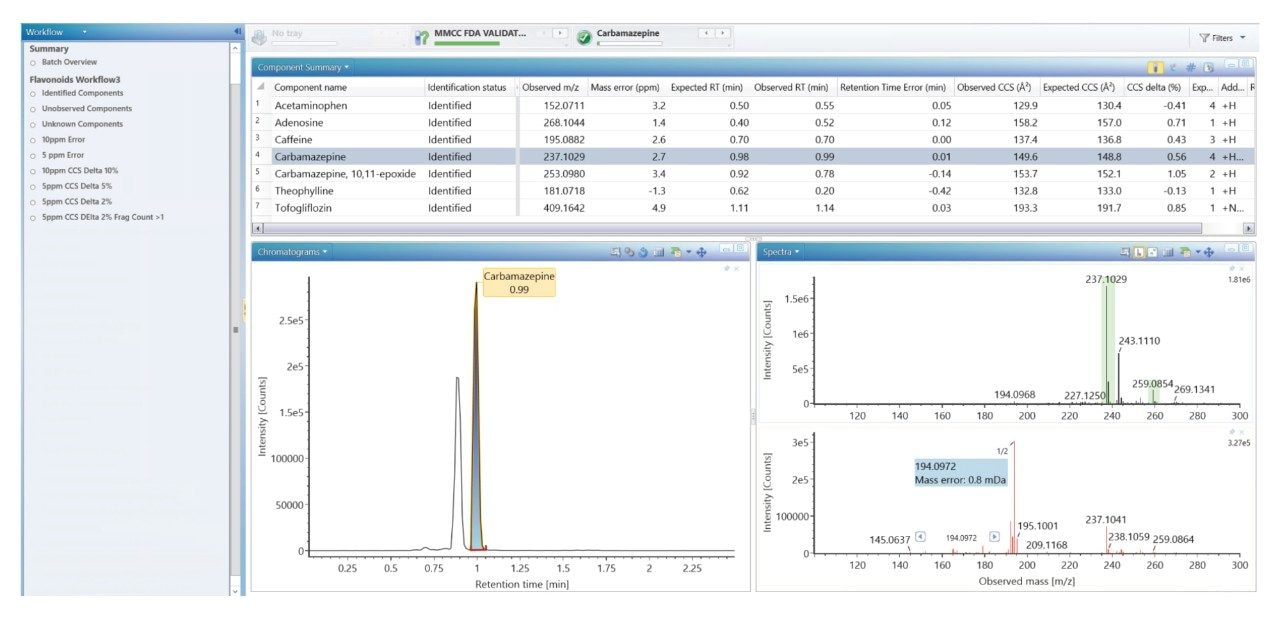
Using a previously described strategy for mass spectrometry library generation,2 a set of FDA-approved small molecule pharmaceuticals were characterized using rapid gradient microbore ultraperformance liquid chromatography ion mobility mass spectrometry (RGM-UPLC-IM-MS). The library generated can be used to facilitate tandem, ToF, or IM based analysis. The strategy employed provides retention time (tr), precursor ion, product ion, and collision cross section (CCS) data for the analytes characterised. A mass spectrometry library has previously been produced using a conventional UPLC method which had a total cycle time of 12 min, where a 2.1 mm × 100 mm column was used, with an applied eluent flow rate of 0.5 mL/min. Here an equivalent rapid gradient microbore UPLC-IM-MS library has been generated, using a rapid microbore metabolic profiling UPLC-MS method, where an eluent flow rate of 0.4 mL/min was employed with a 1 mm × 50 mm column, these conditions reduced the total analysis cycle time to 2.5 min. The conventional and rapid gradient UPLC-MS methods provided peak capacities of 150 and 50 respectively.3
High resolution mass spectrometers (HRMS), such as quadrupole time-of-flight (Q-Tof) mass analyzers, have become more prevalent as screening tools in clinical, forensic toxicology, and metabolite identification research areas.4,5 In non-targeted “full scan” data acquisition, thousands of detections can be made in a single analysis, which is subsequently followed by retrospective targeted data analysis.6 In many areas of research (for example testing for pesticides, mycotoxins, and natural plant toxins), there exists a drive towards higher sample throughput, time efficiency, and cost reduction, which has resulted in movement towards multiclass compound analysis.7 Such a goal, given the common challenge of sample complexity, also exists in metabolic phenotyping which is performed to understand the interactions between genotype, environment and lifestyle, at the molecular level. These investigations may comprise thousands of preclinical metabolism/toxicological, clinical, and epidemiological samples.3
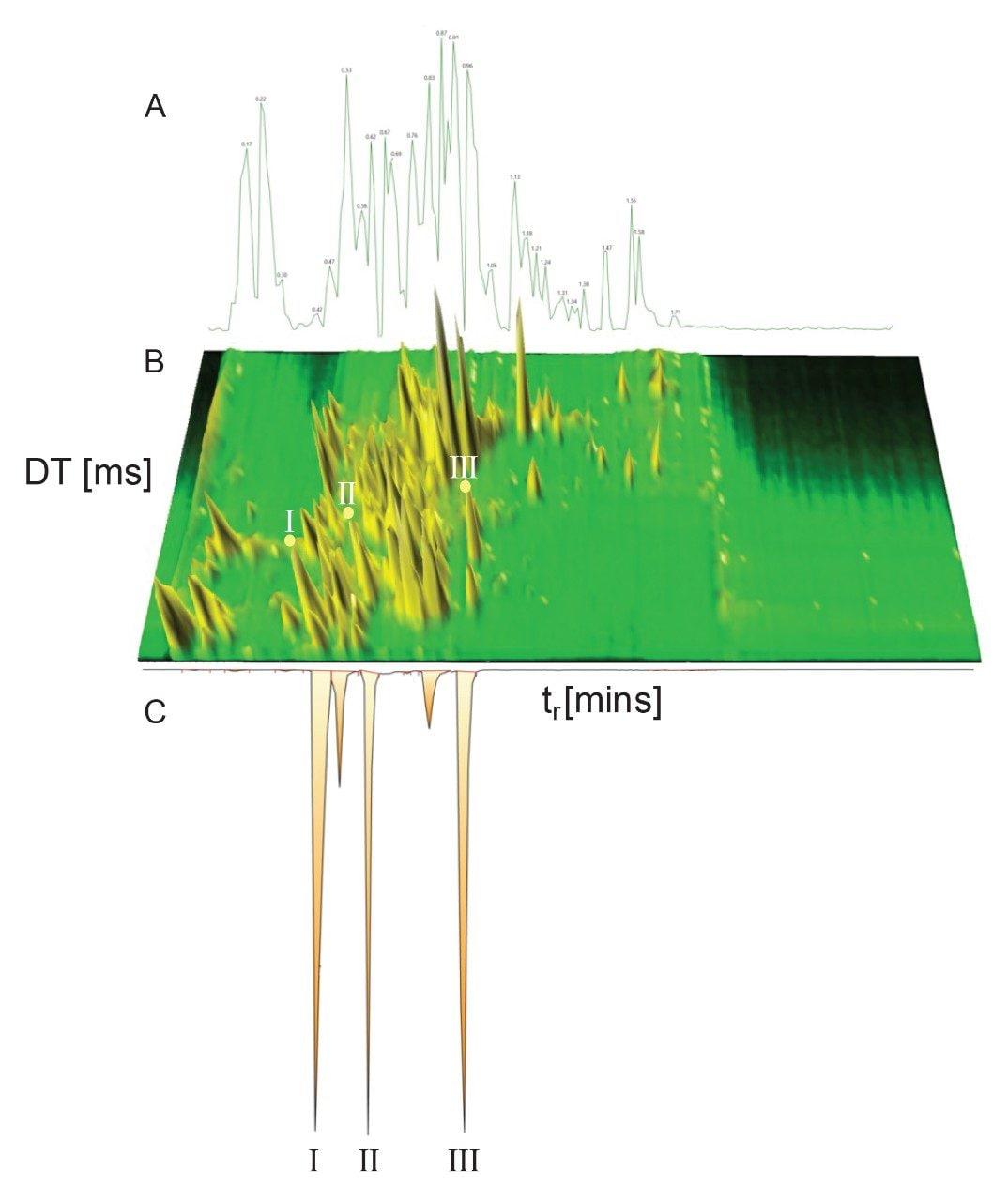
Dwivedi et al discussed the orthogonality of LC, MS, and IM, showing that peak capacity is increased between a factor of 2 to 10, depending on the MS and IM resolution.8 Combining the RGM-UPLC method with ion mobility mass spectrometry affords the opportunity to enhance peak capacity, specificity and analysis flexibility, while retaining the time efficiency of a strategy that provides a 5-fold increase in sample throughput.9,10 UPLC-IM comprises ion mobility (gas phase separation prior to MS analysis) coupled with UPLC (neutral species separation).11,12 The timescale of UPLC (seconds), IMS (milliseconds), and time-of-flight MS (microseconds) are compatible with the requirement of high throughput analysis of complex samples. Ion mobility separation of compounds results from gas phase ions being separated within a gas-filled travelling wave ion mobility (TWIM) RF ion guide of the mass spectrometer, prior to the mass analyzer. Mobility separation is obtained by driving packets of ions through an inert buffer gas (nitrogen) using a relatively weak electric field. The number of collisions between ions and the buffer gas cause drift time differences. The resultant separation is based on the application of repeating DC pulses along the RF ion guide; periodically ions are overtaken by the pulses or waves, where fewer mobile species are overtaken more frequently than higher mobility species. Hence the time to traverse the device is mobility dependent and is governed by factors such as the ion mass, charge, and shape. Ion mobility provides an added dimension of separation to that of LC (hydrophobicity) and MS (m/z), in addition to CCS (collision cross section), a complementary identification metric. The utility of CCS to increase identification specificity has been illustrated across a wide range of applications including metabolite identification, pesticide analysis, mycotoxin screening, medicinal plants speciation studies, and food additives analysis.10,13-17
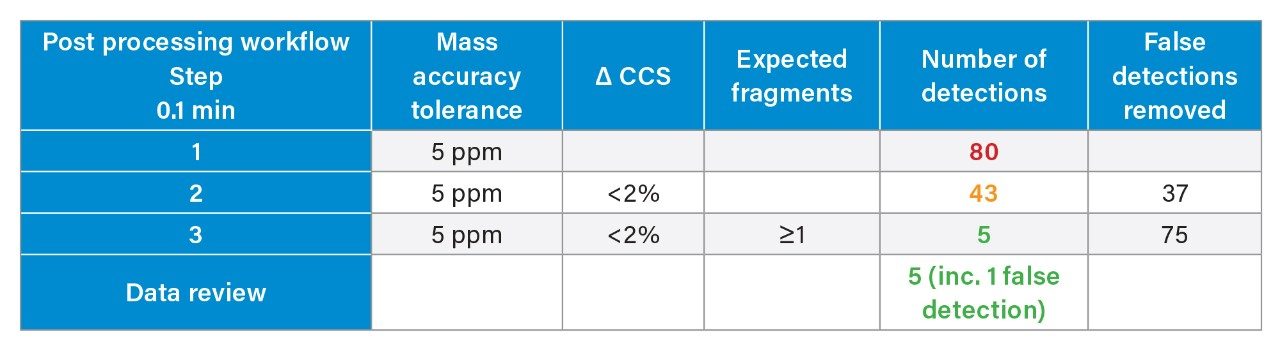
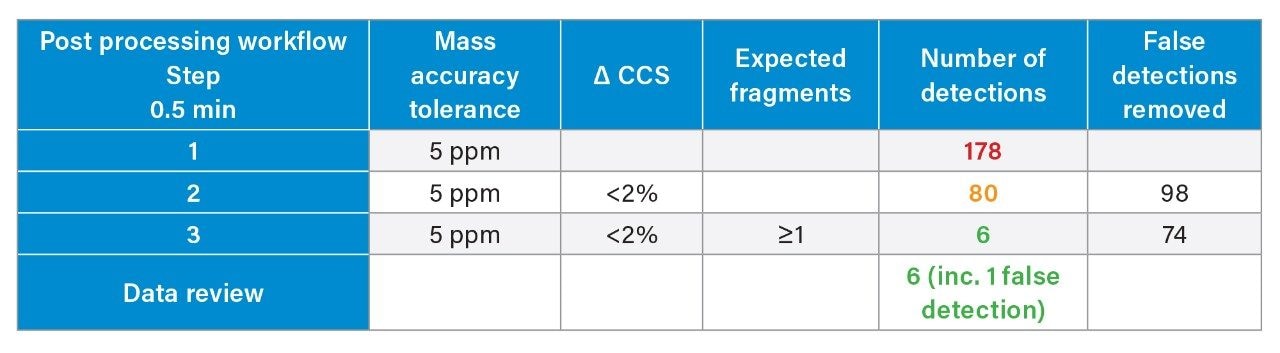
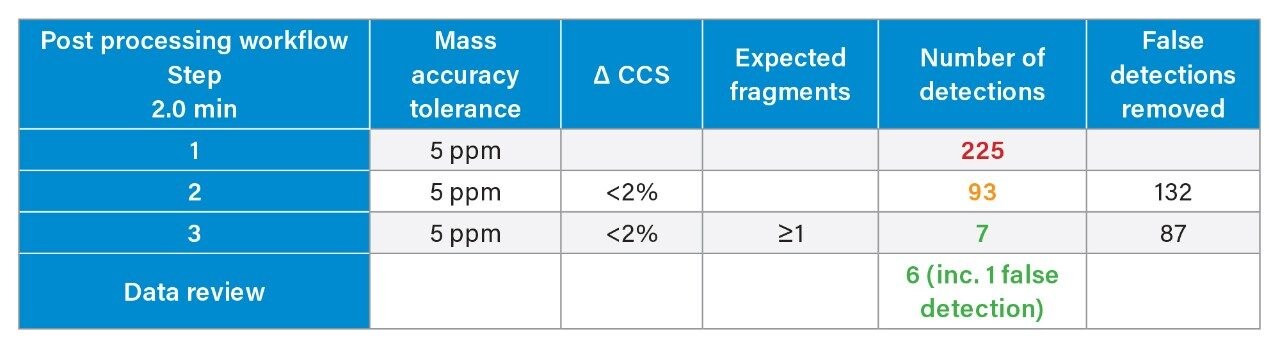
We have used a library of data for a set of small molecule FDA-approved pharmaceuticals to perform a human urine drug screen of a healthy volunteer sample to identify administered pharmaceutical compounds and distinguish them from the endogenous compounds of the complex biological matrix. The feasibility of the approach has been assessed, via comparison of the application of conventional UPLC-IM-MS and RGM-UPLC-IM-MS libraries.