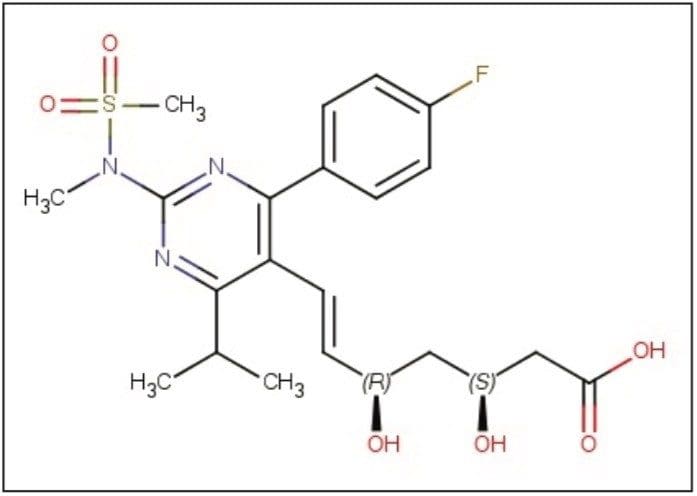
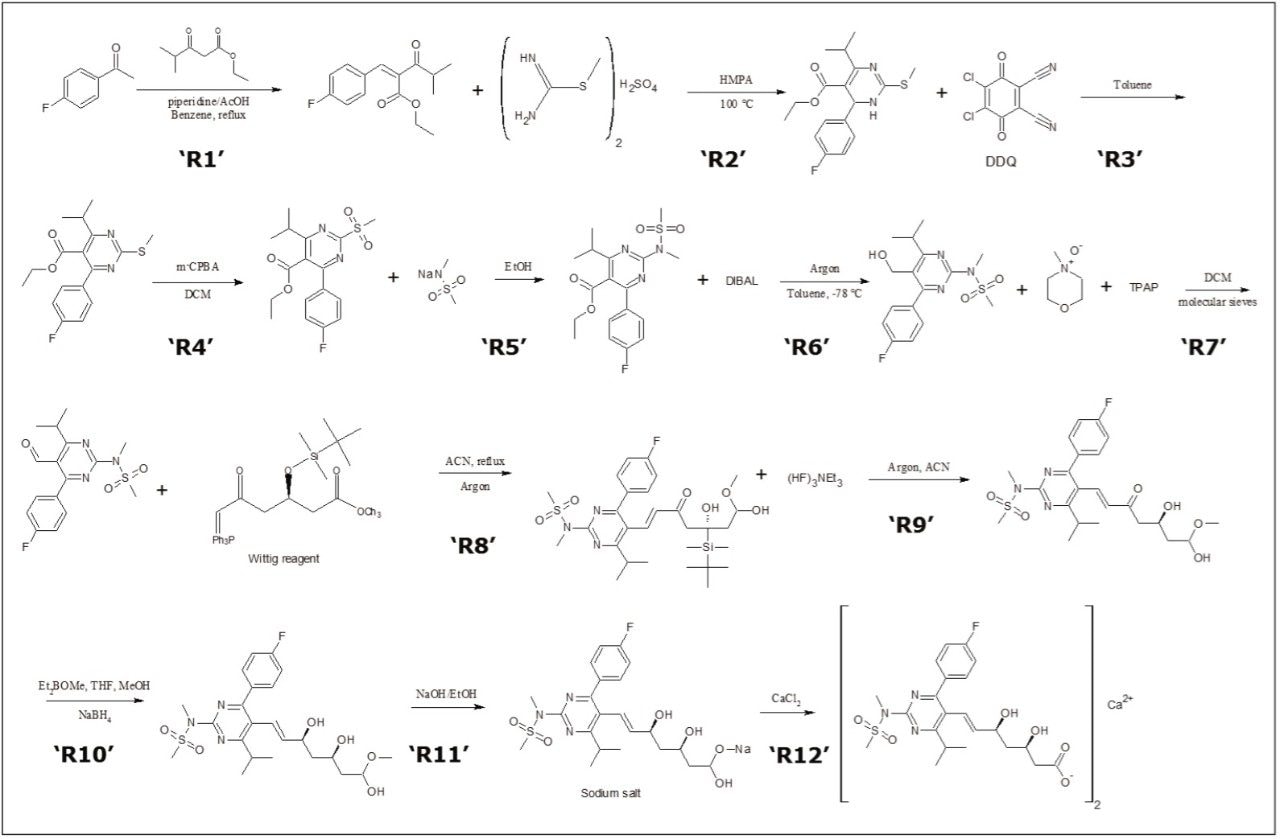
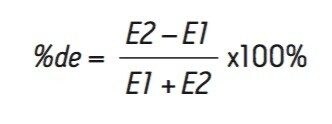Enantiopurity determination
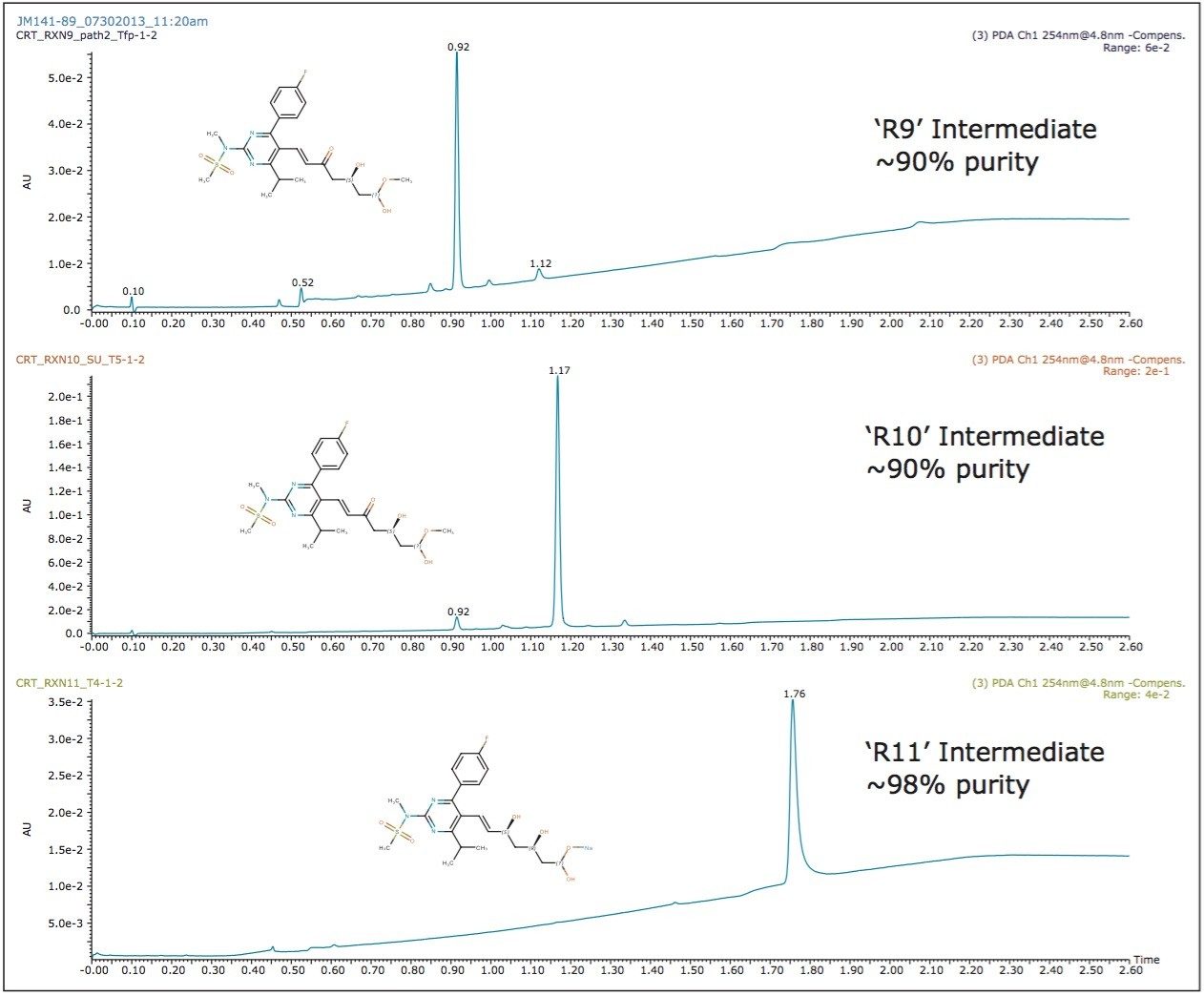
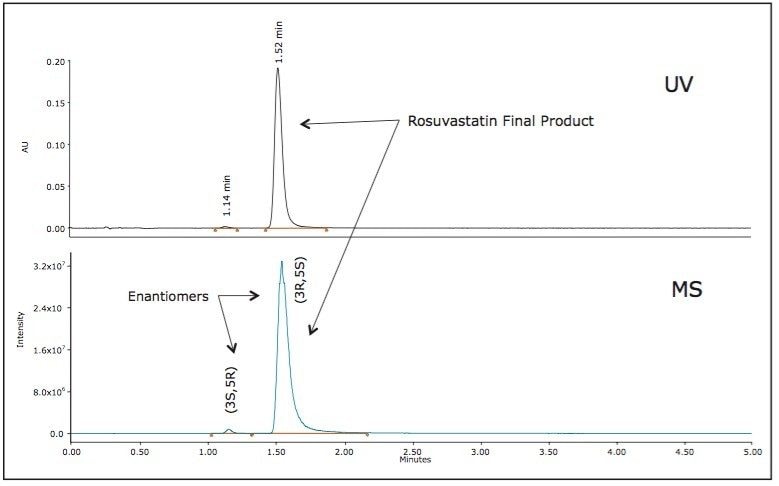
The enantiopurity of the rosuvastatin final product is expected to be relatively high; above 95% purity, based on the reaction scheme described in Figure 2. No major impurity peaks were observed in the chromatographic screening results of the final reactions (R11 and R12), therefore the expected risk of diastereomer impurity interferences in the final rosuvastatin product would be minimal. However, given the stereochemistry of the heptenoic side chain, a variety of diastereoisomer impurities can exist such as (3R, 5R) and (3S, 5S) including the (3S, 5R) enantiomer which requires a chiral stationary phase chromatographic separation. The original patent submission indicates a 10-minute normal phase method utilizing a Chiralpak IB Column yielding a resolution less than 2 to detect the enantiomeric impurity.7 In addition to the poor separation provided by the patented method, the normal phase chromatography is limited due to mass spectrometry incompatibility required for simultaneous identification and confirmation during the chromatographic separation.
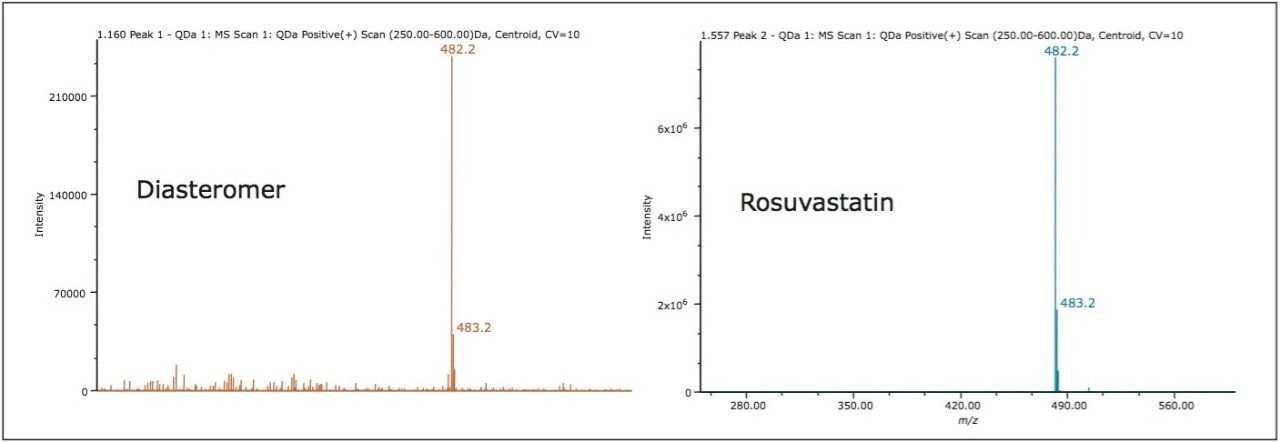
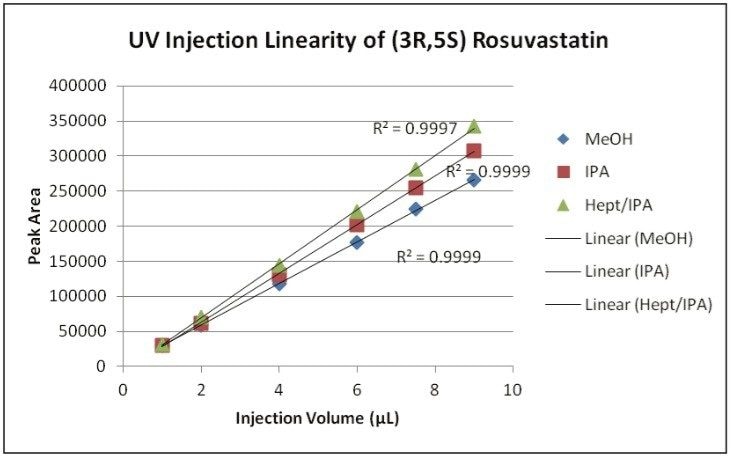
A chiral chromatographic method was created to resolve rosuvastatin enantiomers using an ACQUITY UPC2 Trefoil CEL1, 2.5 μm Column (cellulose tris-(3,5-dimethylphenylcarbamate). The final isocratic method is composed of a mixed alcohol co-solvent with a basic additive. The resolution between the enantiomers was greater than 2.0. The peaks detected by the chiral methodology were confirmed to be rosuvastatin enantiomers (m/z = 482.2 Da) as determined by the ACQUITY QDa Detector. The two diastereomer peaks were integrated and the percent diastereomer excess (%de) was calculated as: