Polyoxymethylene sorbitan monooleate (PS-80) and polyoxymethylene sorbitan monolaurate (PS-20) are common surfactants used in the manufacturing and formulation of biopharmaceuticals. As surfactants they are critical components that aid in minimizing protein denaturing, aggregation and adsorptive losses to surfaces.1 As a stabilizing excipient, knowledge of their composition and stability is necessary to determine shelf-life and safety of drug products used in clinical settings. This is of critical importance as degradation of polysorbate caused by oxidative and/or hydrolytic processes has been linked to reduced efficacy of polysorbate containing drug products.1–2 More recently, enzymatic activity from residual host cell proteins such as lipase/esterase have been identified as an additional degradation pathway of polysorbate further increasing the risk to patient safety.3 To this end, characterization, identification, and quantitation of such excipients must be performed to demonstrate drug products containing polysorbate are safe and efficacious in their formulated state.
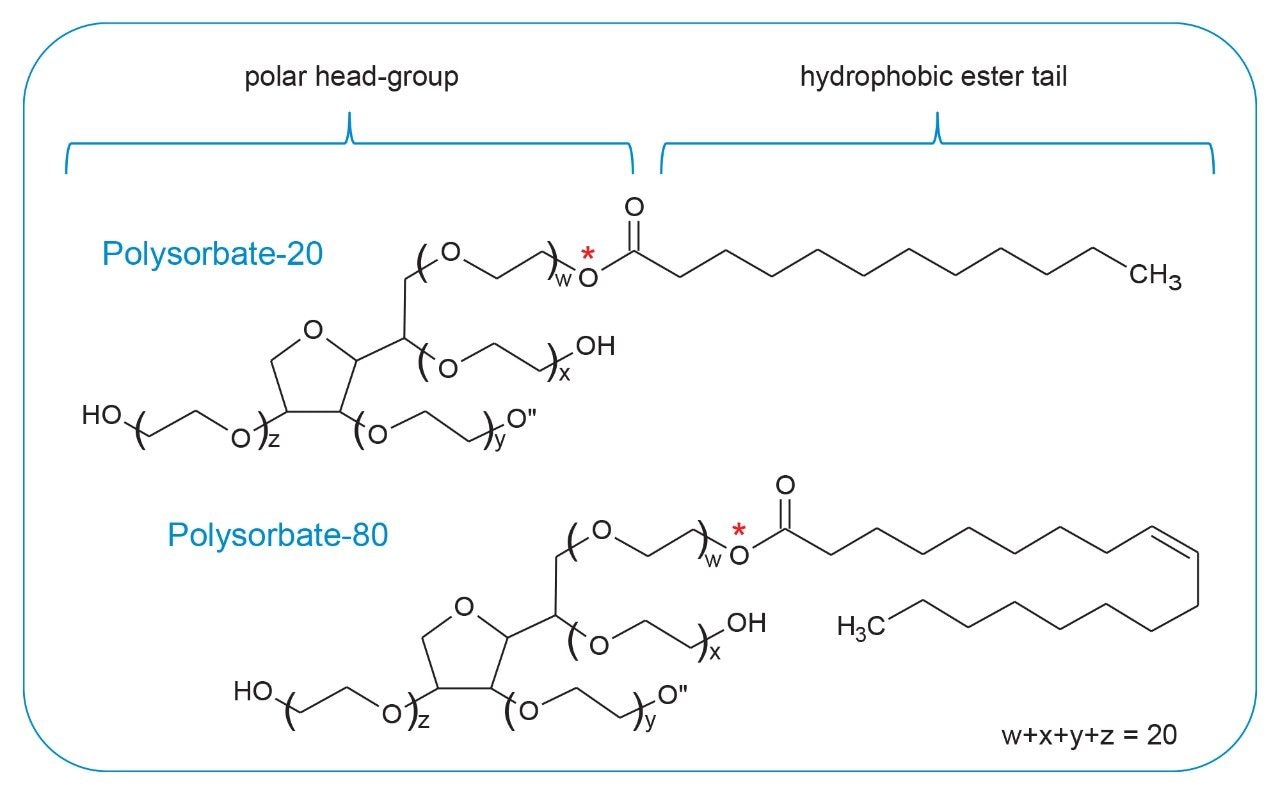
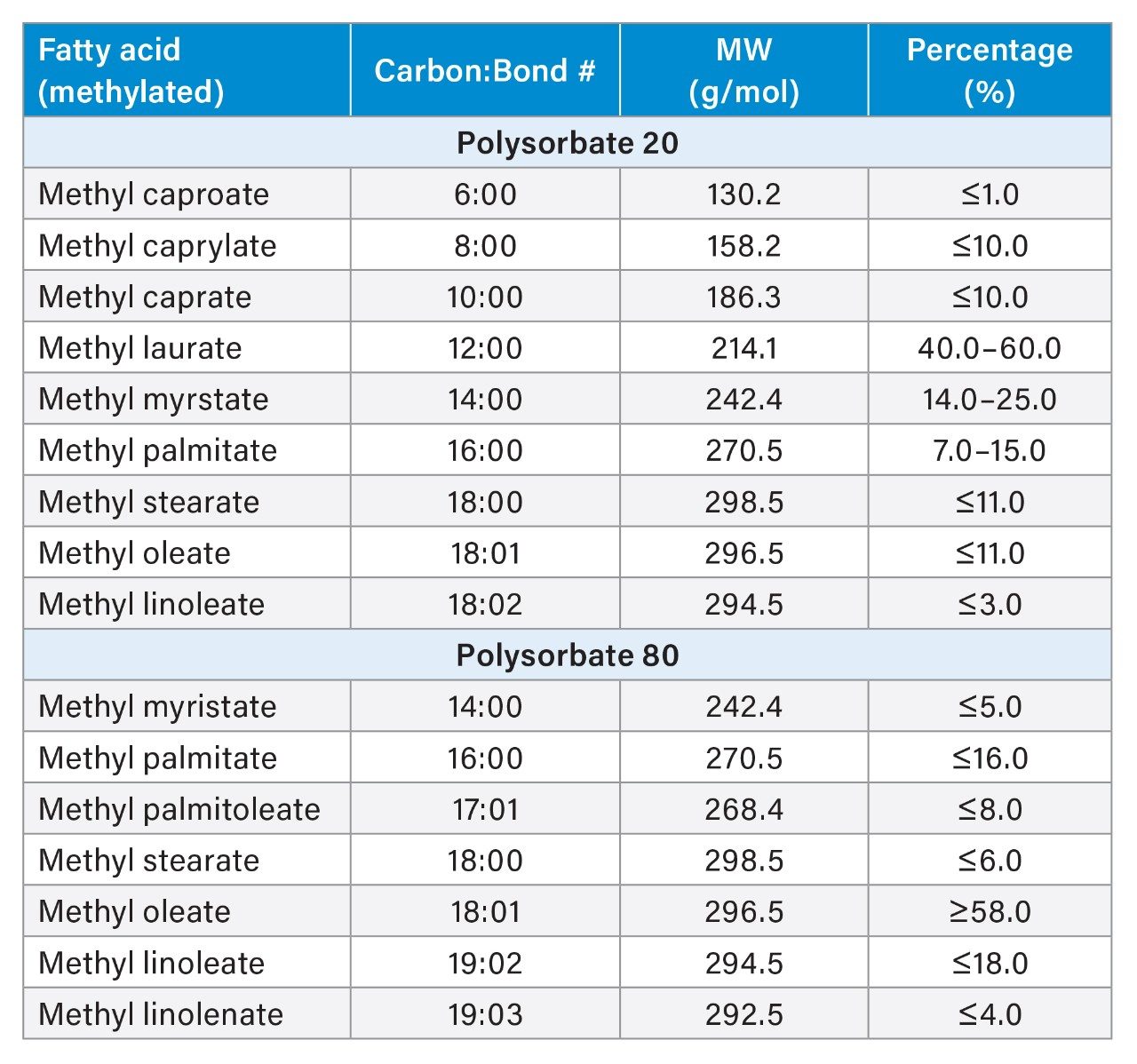
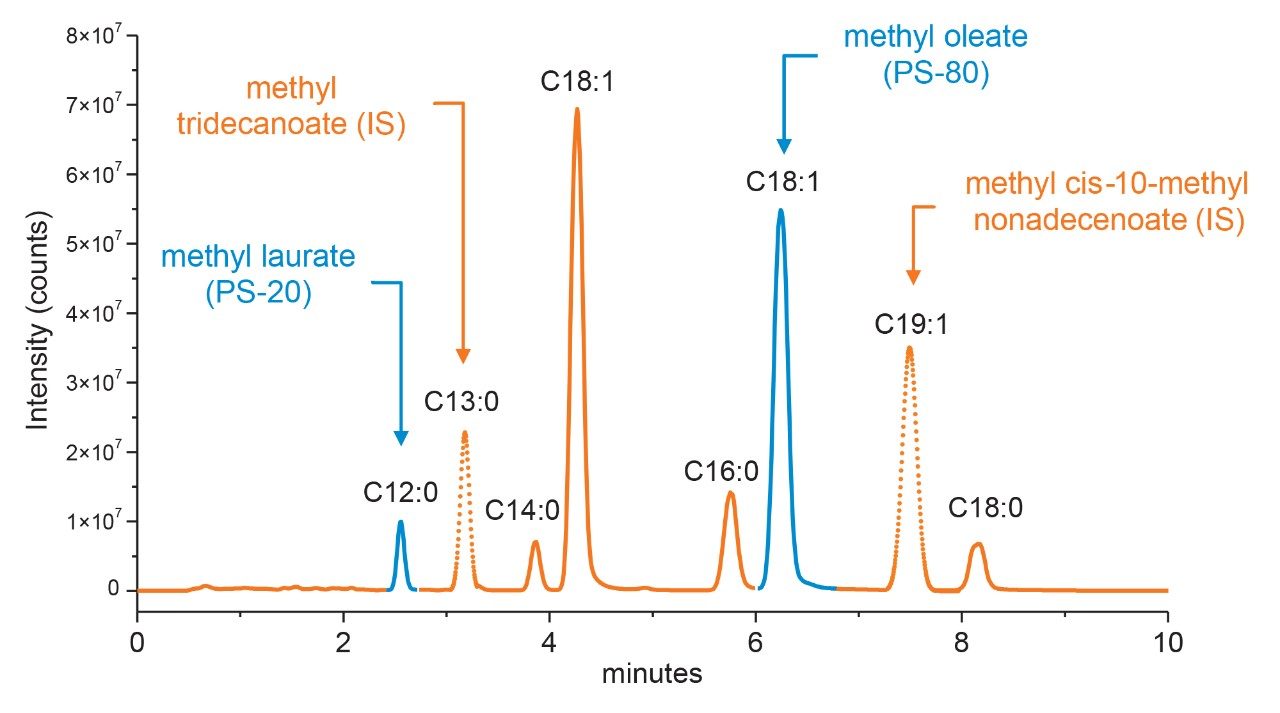
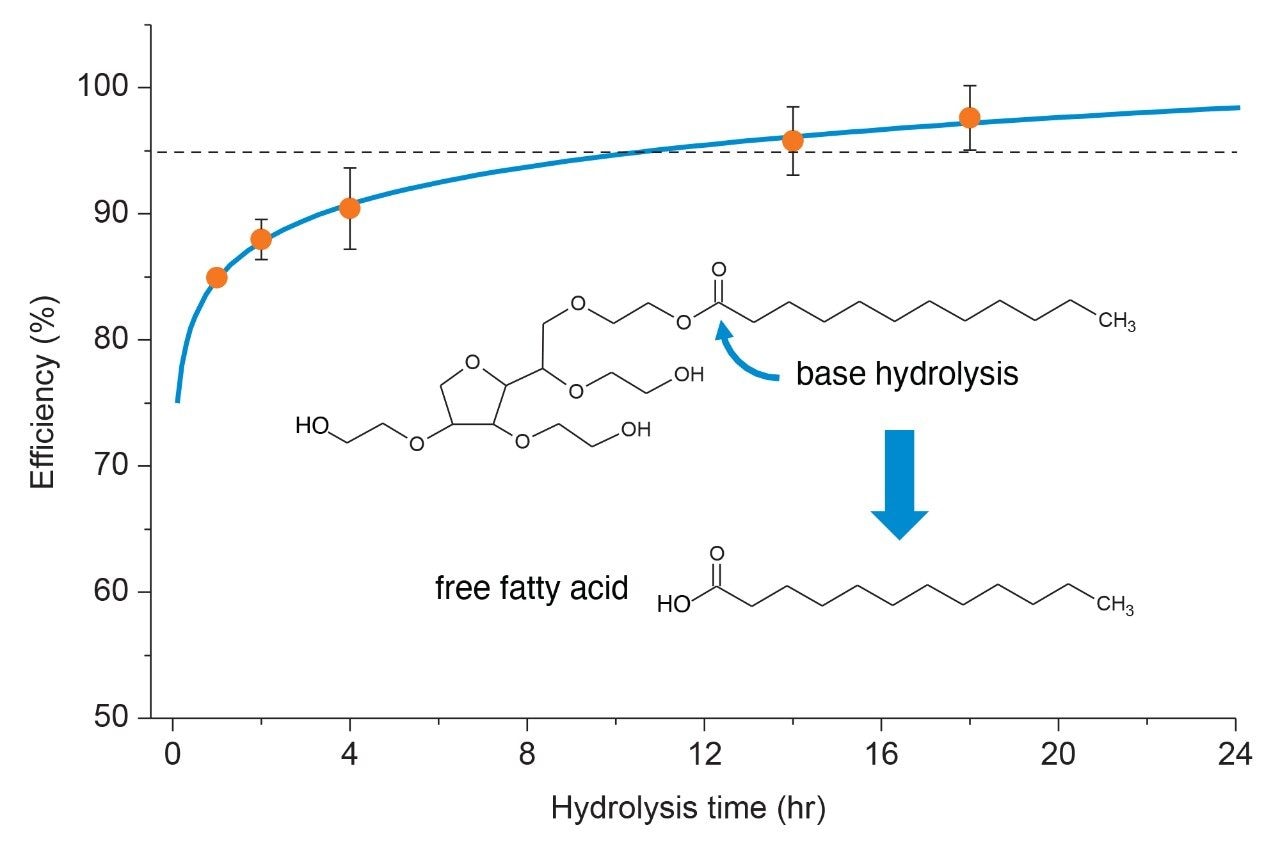
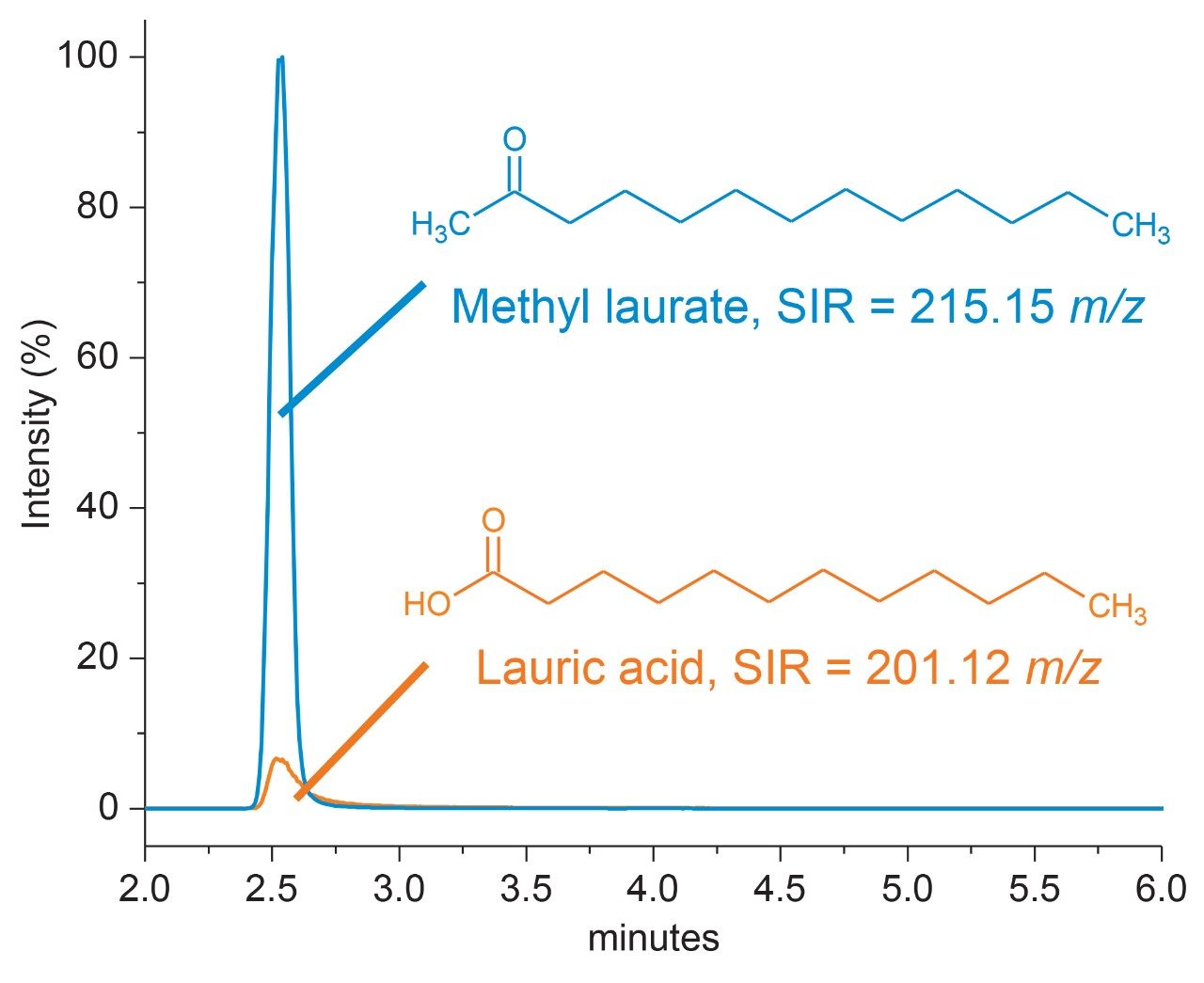
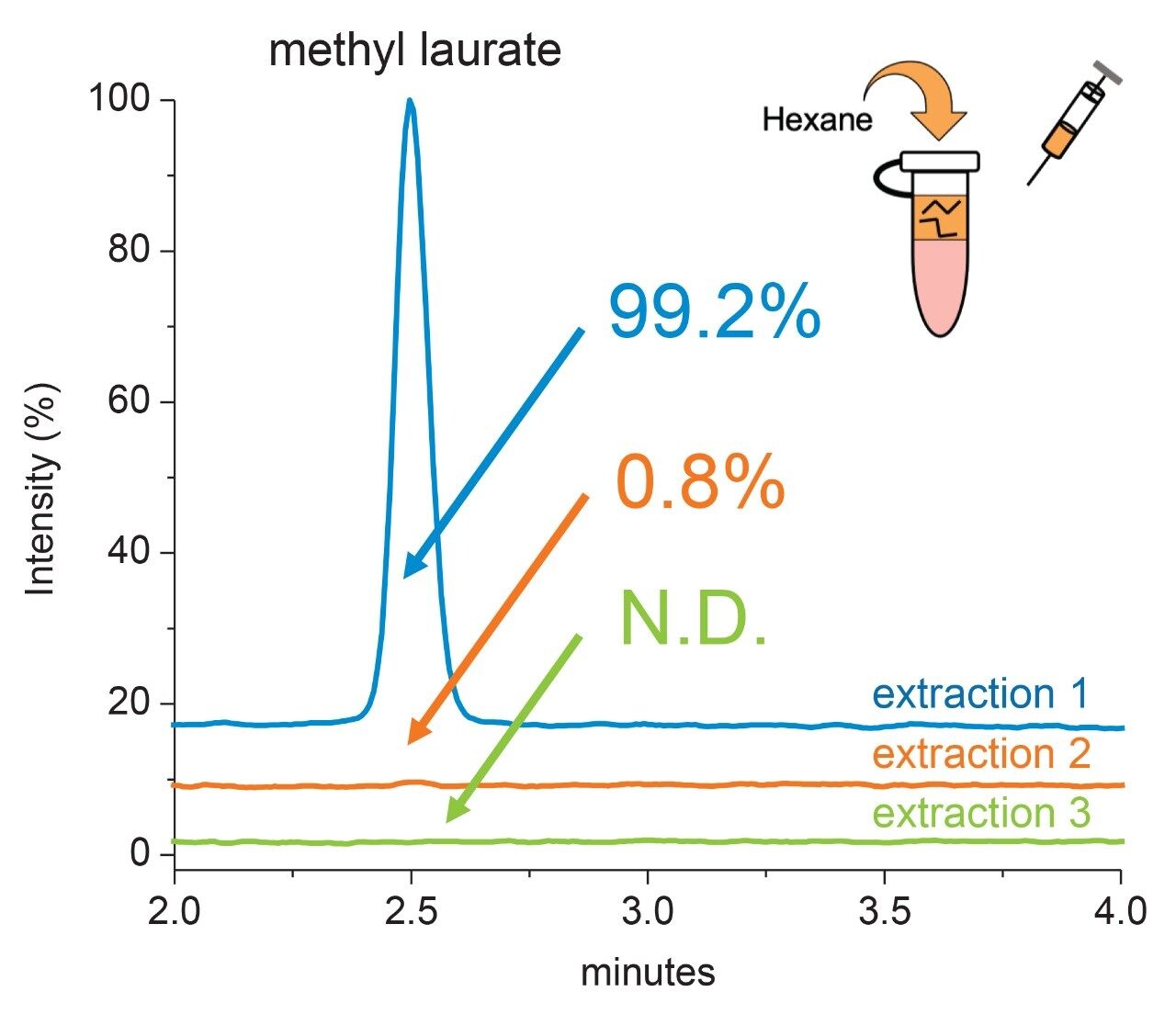
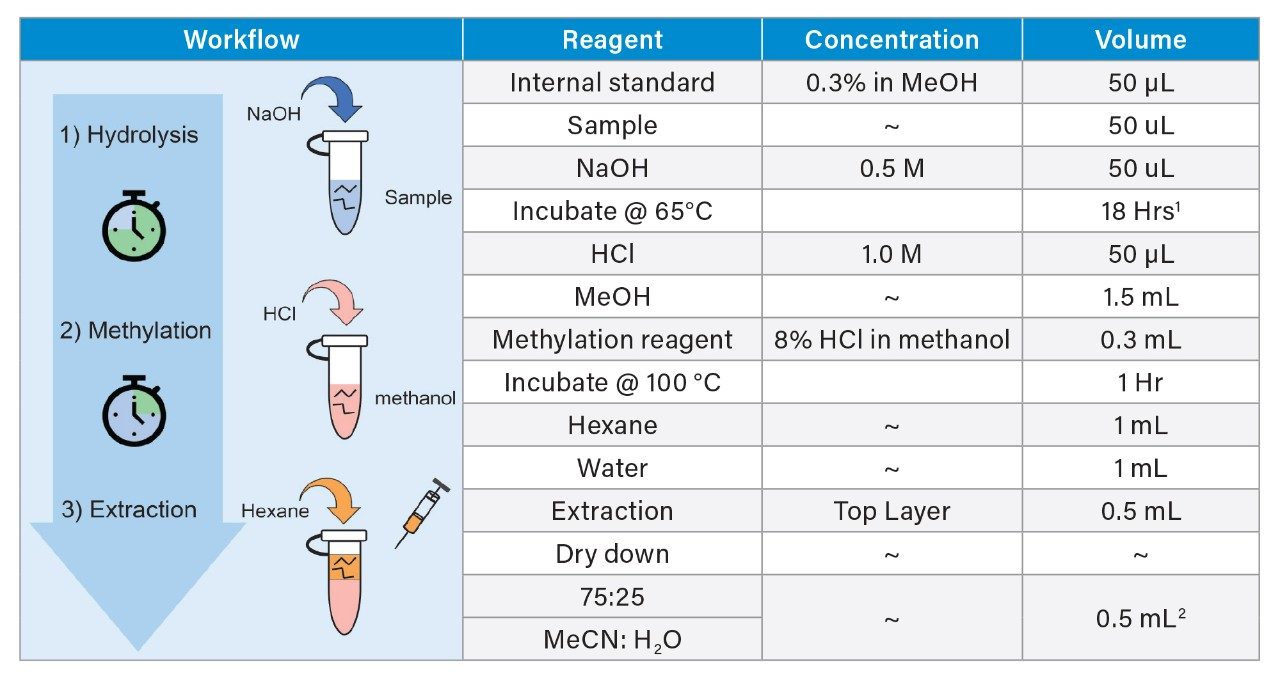
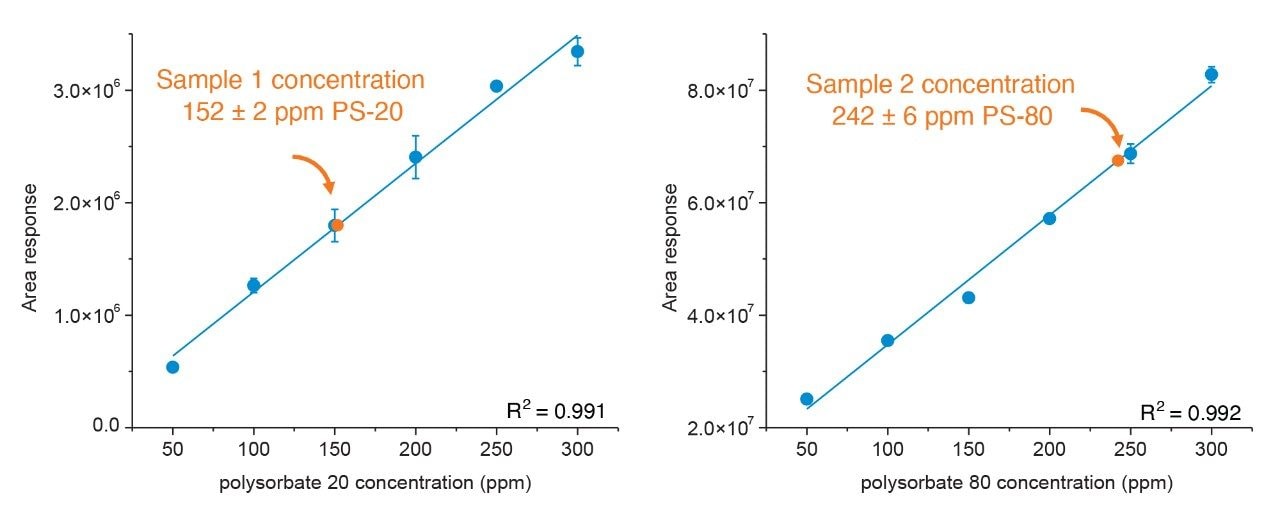
Polysorbate, which is comprised of a polar head-group and hydrophobic tail as shown in Figure 1, is non-volatile in nature and has negligible UV-activity. These unique properties have led to the use of alternative detection techniques such as evaporative light scattering detection (ELSD) and charged aerosol detection (CAD) in the analysis of polysorbate at an intact level.4–5 These methods however, when deployed on HPLC systems, suffer from long elution times (upwards of 40 minutes), as well as complex sample preparations utilizing hazardous materials and reagents. Furthermore, while ELSD and CAD detectors have proven to be useful in the analysis of intact polysorbate, they often lack the fidelity to detect subtle changes in polysorbate concentration and are limited to ppm-levels of detection. Recently, we published a method for the analysis of polysorbate 80 based on common practices found in literature that was shown to be efficient and sensitive.6 In this method, as depicted by the workflow shown in Figure 2, it was demonstrated that sub-ppm levels of detection could be achieved using UV-based detection for the hydrophobic fatty acid tail (oleic acid) following a base hydrolysis of the polysorbate 80 at the ester linkage noted by the asterisks in Figure 1. This method, which used isocratic conditions, relies in part on the notion that the polysorbate raw material is relatively pure with respect to composition and the free fatty acid of interest is UV-active. In practice, as shown in Table 1, polysorbate can be comprised of multiple fatty acid esters conjugated to the base sorbitan molecule each of which can have varying levels of UV-activity based on the structure (saturated vs. unsaturated) of the associated alkyl tail. This is further compounded by the fact that the fatty acid profile of the polysorbate types can span a broad range in concentration increasing the challenge to developing a platform-like UV-based method that can provide sufficient detector response for target constituents of polysorbate in biopharmaceutical samples. Given this, a method that offers increased specificity and sensitivity towards polysorbate and one that can be universally deployed in the analysis of PS-20 and PS-80 containing drug products is highly desirable.