An Efficient LC/MS Workflow for Identification and Monitoring of Host Cell Proteins for Assisting Monoclonal Antibody Process Development
Abstract
This application note introduced two analytical workflows for LC-MS analysis of host cell proteins (HCPs) in monoclonal antibodies: HCP Discovery and HCP Monitoring. The HCP Discovery workflow was demonstrated by identifying HCPs down to 5 ppm in the NIST mAb Reference Material, using the bench top Xevo™ G3 QTof System based on analytical scale UPLC™ separations of digested proteins. The HCP Monitoring assays were performed on the BioAccord™ UPLC-MS System, a platform fundamentally designed for high performance routine operation by non-MS experts, also using the compliance-ready waters_connect™ software for data acquisition and processing.
Benefits
- A Discovery HCP assay using UPLC-QTof MS, capable of detecting low-level HCPs, down to a concentration of 5 part-per-million (ppm) in the NIST mAb Reference Material 8671
- An HCP Monitoring assay, developed on a platform ideal for non-MS experts, was able to achieve the same detection limit (5 ppm) for a protein digest standard spiked into the NIST mAb digest
- Both Discovery HCP and Monitoring HCP workflows incorporate an analytical scale separation on the ACQUITY™ Premier UPLC System and Premier CSH™ Ccolumn chemistry that improves overall robustness for peptide based analysis
Introduction
Host cell proteins (HCPs) are endogenous, process-related impurities derived from expression systems during biotherapeutic production. Even at trace levels (0.1–100 ppm), these impurities have the potential to produce unwanted immunogenic response in patients, or can reduce the safety, efficacy, or stability of the therapeutic.
For these reasons, the regulatory agencies require manufacturers to demonstrate clearance and control of HCP impurities during product purification to show process understanding and ensure acceptably low HCP levels for clinical studies. This requires analytics enabling the establishment of HCP identity and assays capable of individual and/or aggregate HCP quantification. In early development, high capability mass spectrometry systems, such as Waters™ Cyclic™ IMS and SYNAPT™ HDMS systems are used for HCP Discovery, employing complex proteomics-like workflows and operators with significant LC-MS expertise. As the drug development progresses, it is desirable to use much simpler, more routine immunological assays and LC-MS analysis workflows, to progress molecules through process development and into commercial production.
The biopharmaceutical industry has traditionally relied on ELISA assay for measuring the total HCP concentration, but mass spectrometry-based HCP analysis has emerged in recent years as a powerful alternative, providing more extensive (proteome-wide) HCP coverage and is able to further reduce risk by measuring individual HCP levels. 1–11
The composition of HCPs from the NIST mAb Reference Material 8671 has been thoroughly investigated using a variety of sample enrichment protocols.5–8 Here we adapted a mAb depletion protocol, originally introduced by Eli Lilly, for enzymatic digestion of NIST mAb HCPs, using a novel recombinant trypsin from Waters Corporation – RapiZyme.5,12
This application note is focused on method optimization for routine HCP analysis using analytical scale UPLC separations coupled with either QTof or Tof MS. Two workflows are discussed here: 1) Discovery (ID-enabling) and subsequent quantification of unknown HCPs using the NIST mAb Reference Material; 2) Monitoring of known HCPs using the NIST mAb RM digest as sample background spiked with known concentrations of protein digest standards.
The first workflow described here is based on the HCP Discovery Assay performed in MSE mode (DIA) using extended (90 minute) peptide gradient separations. The HCPs are identified by a proteome-wide database search following data processing with Progenesis QI for Proteomics (QIP) 4.2 software. The MSE results can be assembled into spectral libraries, containing the identified peptide precursors, observed charge states and apex retention times. A popular third-party proteomics software, Byonic (Protein Metrics), was also used to process the Discovery HCP data, to demonstrate the data compatibility with a vendor-agnostic software. In addition, this also served the purpose of independently verifying the HCP assignment for Progenesis QIP.
The second workflow is more typical for targeted monitoring of known HCPs from mAb samples that are collected across various purification stages. A higher–throughput HCP Monitoring Assay employing MSE data acquisition with 30 minute peptide separations was developed on a small foot print, ease-of-use BioAccord UPLC System. Data processing was performed using the Accurate Mass Screening workflow in waters_connect, specifically designed for targeted analyte monitoring. This HCP monitoring processing method is embedded in a compliant ready software – waters_connect, for quantification and monitoring of all identified HCPs across many samples (10–50) resulted from biopharmaceutical purification using different purification protocols. In this application note, the HCP workflow described above was applied to the identification and monitoring of HCPs and spiked MIX-5 protein digest standards in the NIST monoclonal antibody.
Experimental
Sample Preparation
A highly purified mAb (NIST mAb candidate reference material LRM 8671, 10 mg/mL concentration) produced in a murine cell culture was purchased from Millipore Sigma (St Louis, MO). The NIST mAb was digested using a protocol designed to remove the high-abundance mAb species through protein precipitation, using a modified version of a previously published digestion protocol.5 In a 500 µL Protein LoBind Eppendorf tube (catalogue no 022431064), 200 µL of NIST mAb were mixed with 20 µL of 1 M Tris HCl buffer (containing 1 M CaCl2) and 10 µL of 4 µM RapiZyme trypsin (Waters p/n: 186010107) and digested overnight (~ 16 hours) at 37 °C. As previously described, the digestion protocol avoids mAb denaturation, reduction, and alkylation to favor the digestion of HCPs over the digestion of the more stable NIST mAb, kept in a native (non-denatured) conformation.5 After 16 hours of enzymatic digestion, the undigested mAb was denatured by heating and reduction with 4 mM DTT (90 °C, 15 minute) to effect precipitation. The sample was centrifuged for 5 minutes at 12,000 g and the supernatant (~ 200 µL digest) was recovered and acidified with 1 µL of formic acid (Millipore Sigma).
For the HCP Discovery Assay, performed in LC-MSE mode on the Xevo G3 QTof, four protein digest standards (MIX-4: ADH—yeast alcohol dehydrogenase, BSA - bovine serum albumin, ENL – yeast enolase and PHO - rabbit phosphorylase b), were spiked post-digestion in the NIST mAb digest.
For the HCP Monitoring Assay, performed in LC-MSE mode on the BioAccord LC-MS System, five protein digests (MIX-5: ADH, BSA, ENL, PHO and CLP-B—chaperone Ecoli digest) were spiked post digestion in the NIST mAb at various concentration levels (see Figure 6 inset).
LC-MS grade acetonitrile (ACN) was purchased from Thermo Fisher Scientific (Waltham, MA). LC-MS grade water and methanol were ordered from Honeywell (Charlotte, NC).
LC Conditions
|
LC system: |
ACQUITY™ UPLC Premier BSM |
||
|
Column: |
ACQUITY Premier CSH 2.1 x 150 mm, packed with 1.7 µm CSH C18 particles (p/n: 186009462) |
||
|
Column temperature: |
60 °C |
||
|
Flow rate: |
200 µL/min |
||
|
Mobile phases: |
Solvent A: 0.1% FA in LC-MS grade water Solvent B: 0.1% FA in LC-MS grade acetonitrile (ACN) |
||
|
Injection volumes: |
50 µL (HCP Discovery Assay) and 20 µL (HCP Monitoring Assay) Fixed Loop injection using a 100 µL injection loop |
||
|
Wash solvents: |
Purge solvent: 50% MeOH Sample Manager wash solvent: 50% MeOH |
||
MS Conditions (Discovery HCP Assay):
|
MS system: |
Xevo G3 QTof Mass Spectrometer |
|
|
Ionization mode: |
ESI+ |
|
|
Capillary voltage: |
2.8 kV |
|
|
Cone voltage: |
25 V |
|
|
Source offset: |
60 V |
|
|
Source temperature: |
120 °C |
|
|
Desolvation temperature: |
450 °C |
|
|
Cone gas flow: |
50 L/h |
|
|
Desolvation gas flow: |
500 L/hr |
|
|
TOF mass range: |
400–2000 (MSE acquisition) |
|
|
Acquisition rate: |
0.5 sec |
|
|
Low energy CE: |
6 V |
|
|
High energy CE ramp: |
15 to 45 V |
|
|
Data acquisition software: |
UNIFI™ 3.0.0.21 |
|
|
Data processing software: |
Progenesis QI for proteomics v 4.2 Protein Metrics Byonic v 5.2.31 |
|
|
|
MS conditions (Monitoring HCP Assay)
|
MS system: |
BioAccord LC-MS System |
|
|
Ionization mode: |
ESI+ |
|
|
Capillary voltage: |
1.5 kV |
|
|
Cone voltage: |
40 V |
|
|
Source offset: |
30 V |
|
|
Source temperature: |
120 oC |
|
|
Desolvation temperature: |
450 oC |
|
|
TOF mass range: |
50–2000 (MSEacquisition) |
|
|
Acquisition rate: |
0.5 sec |
|
|
Low energy CV: |
40 V |
|
|
High energy CV ramp: |
60 to 80 V |
|
|
Data acquisition software: |
UNIFI 2.1.2.4. |
|
|
Data processing software: |
UNIFI 2.1.2.4 |
Gradient Table: HCP Discovery Assay

Gradient Table: HCP Monitoring Assay

Gradient conditions used for the HCP Discovery assay (A) and for the HCP Monitoring assay (B).
Results and Discussion
HCP Discovery Workflow
Two independent workflows are proposed in the current application note for LC-MS analysis of HCPs. One of the first tasks for HCP analysis is their identification, as each biopharmaceutical protein is expected to have a unique HCP composition related to the purification protocol employed to purify it.
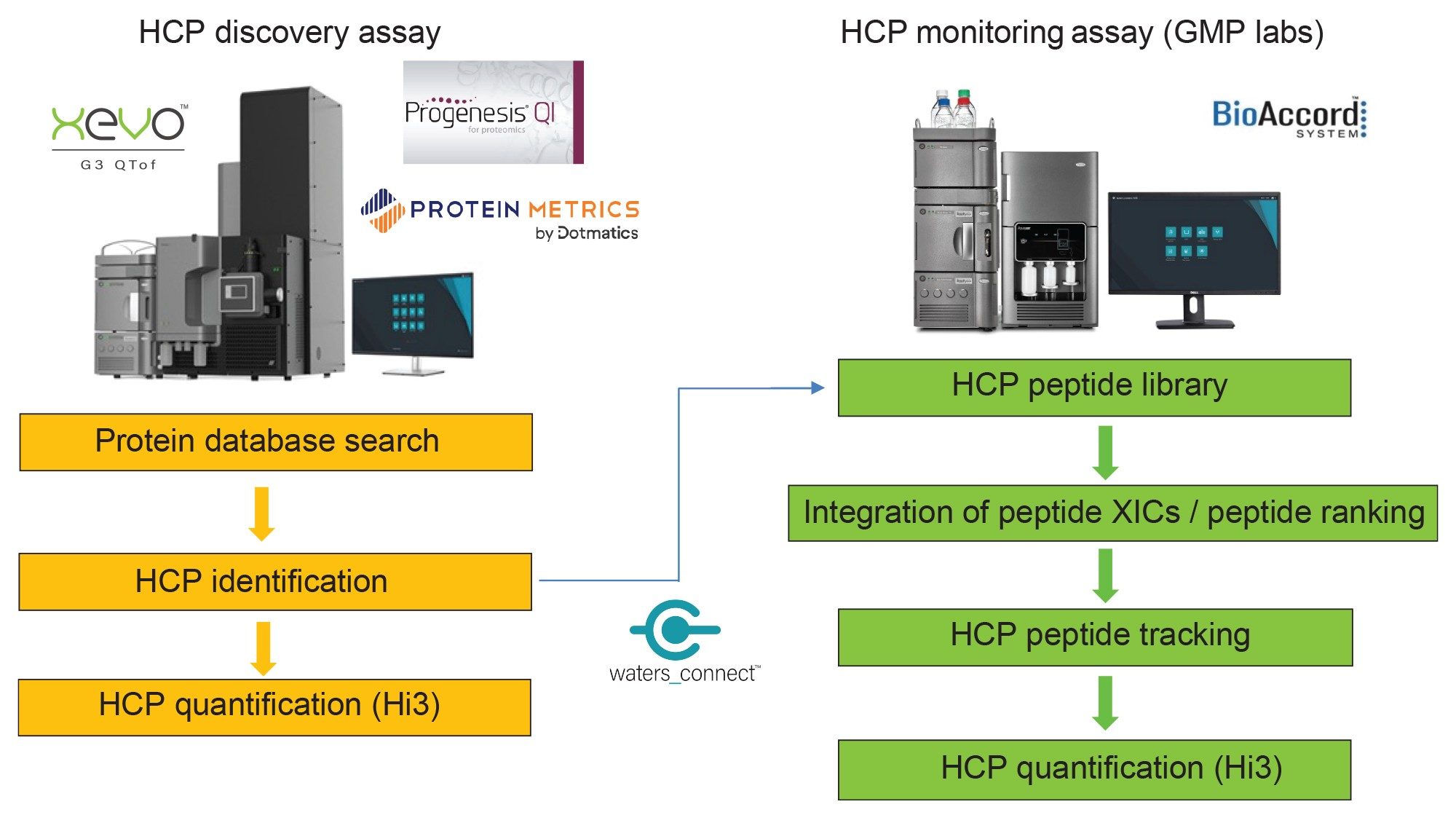
As illustrated by the Figure 1 flow chart, the HCP Discovery Assay is responsible for identifying each of the (previously unknown) HCP contaminants. Only LC-MS assays can provide this specific information, while ligand binding assays (ELISAs) are unable to identify individual process-related contaminants. In addition, the HCP Discovery Assay provides quantification information for each HCP. Once all the HCPs are identified down to a specified threshold (typically expressed relative to the very abundant biotherapeutic protein, e.g. a level of 10 ppm), the HCPs can be tracked across multiple biopharmaceutical samples produced by different cell culture conditions or purification protocols.
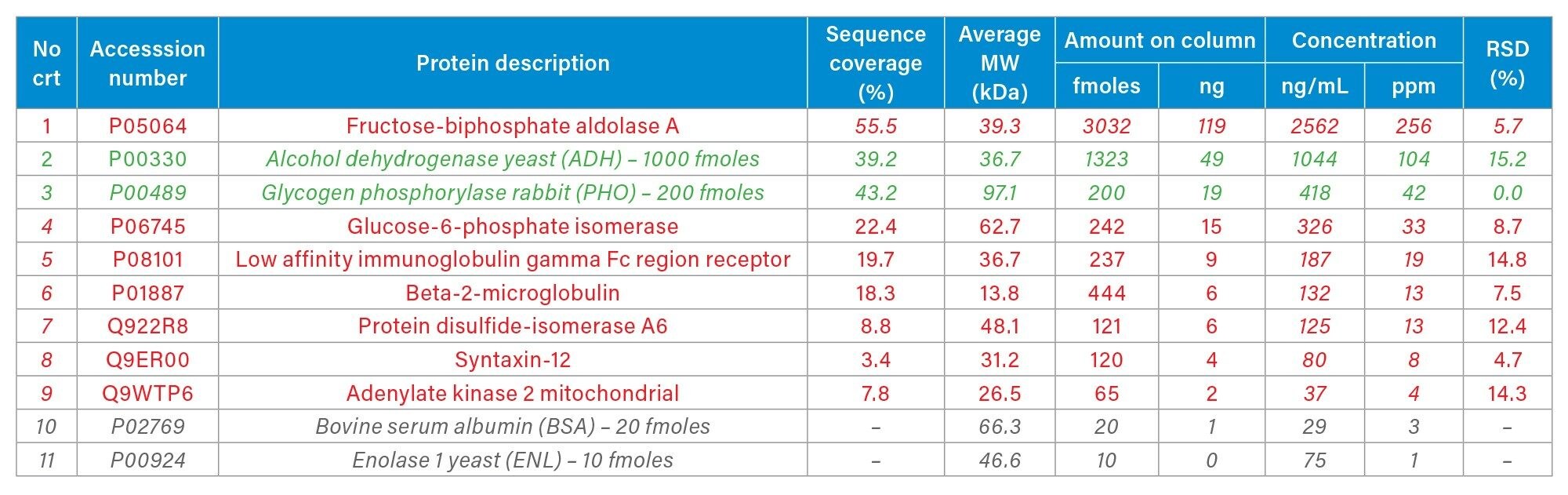
HCP impurities contained in the commercially available NIST mAb (Reference Material 8671) have been previously characterized, with fructose biphosphate aldolase A being the most abundant protein, present in the concentration range of 200-300 ppm. 1,2,5–9 The base-peak chromatogram recorded for the separation of the NIST mAb digest, under the experimental conditions of the Discovery HCP Assay (90 min gradient, high sample load ~50 µg), is displayed in Figure 2. This one-dimensional UPLC-MSE assay was able to identify six low-abundance HCPs, present in the concentration range of 40 down to 5 ppm, in addition to higher-abundance aldolase (Table I).

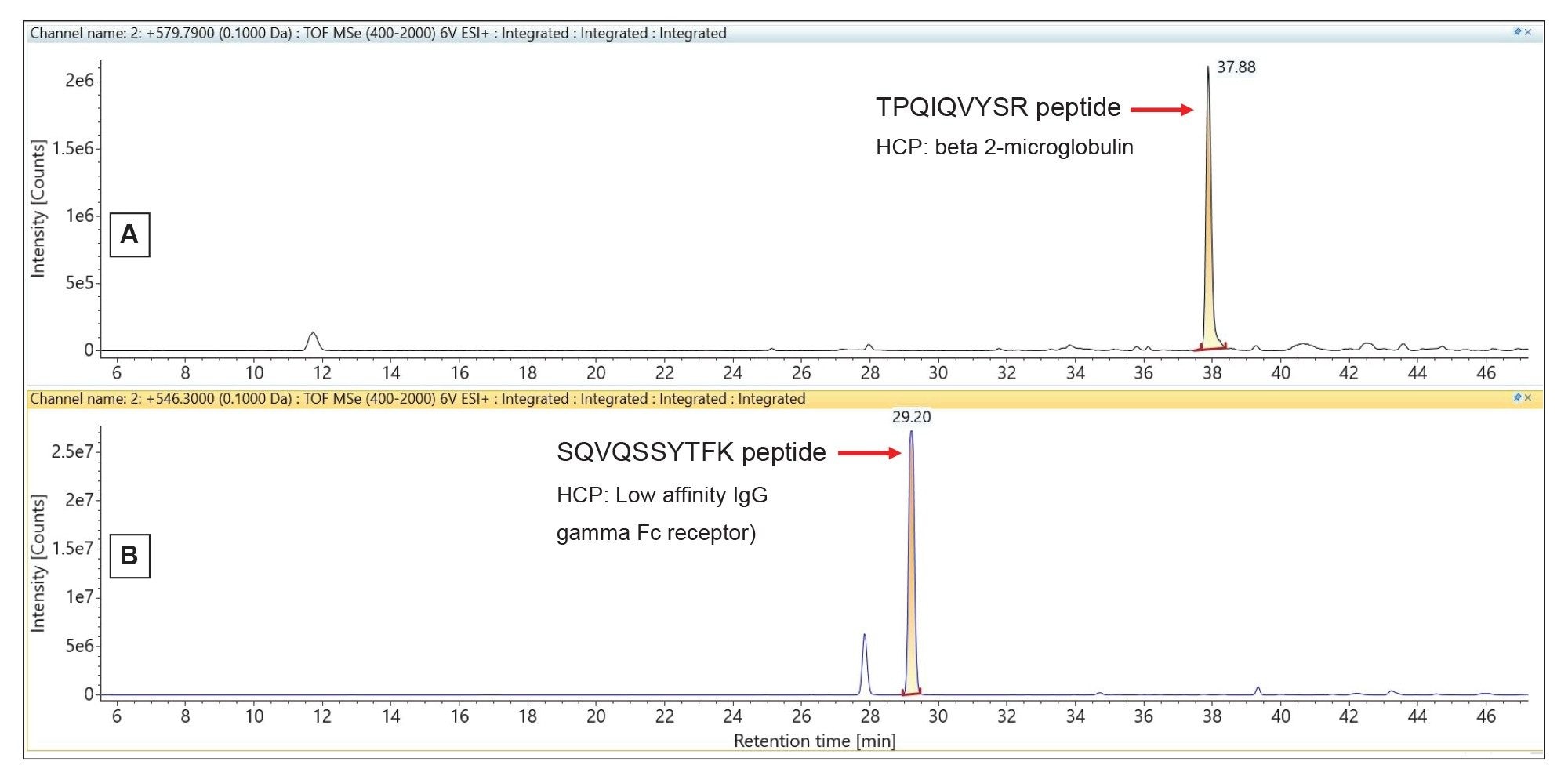
The extracted mass chromatograms of two peptide precursors, one from beta-2-microglobulin (with a measured concentration of 13 ppm) and the other from Low affinity IgG gamma Fc receptor protein (19 ppm measured concentration) are shown in Figure 3. The Premier CSH column, specifically designed to maintain good chromatographic resolution under column overloading conditions, provides symmetrical peak shapes (~20 sec peak width at 10% of peak height) for all low-abundance HCP peptides, despite the high amount of mAb digest loaded on-column (50 µg).
The NIST mAb digest was analyzed in triplicate and each dataset was searched against the mouse proteome database (16,644 protein entries) using two different software platforms: Progenesis QIP and Byonic (Protein Metrics). A screenshot containing the Progenesis QIP results obtained for the analysis of one digest replicate is displayed in Figure 4. Proteins also identified by the Byonic software for the same injection are highlighted with the asterisk symbol (*). The HCP identification results obtained from these two search engines, operating with very different protein identification algorithms, were in very good agreement, with seven low-abundance NIST mAb HCPs identified by both (Table I).

The NIST mAb HCPs along with the MIX-4 spiked proteins were quantified utilizing the “Hi3” approach.13 The “Hi3” protein quantification is based on finding the top three peptides from each protein that produce the best MS1 response following ESI-MS ionization. The ESI-MS response for these three best ionizing peptides is then summed and compared against the response of a spiked protein with a known concentration - in this case PHO protein spiked at 42 ppm (Table I), in order to calculate the concentration of all detected NIST mAb HCPs.
The complete list of HCPs identified in two out of three replicates using the Progenesis and Byonic results is displayed in Table I. Overall seven NIST mAb HCPs were identified and quantified. The lowest concentration measured was 4 ppm for adenylate kinase 2, indicating the ability of the one-dimensional Discovery HCP Assay to identify unknown HCPs in the low-ppm range (down to ~5 ppm) in consistent fashion.
HCP Monitoring Workflow
The HCP Monitoring Assay, highlighted by a separate flowchart in Figure 1, is responsible for detecting multiple HCP peptides in higher-throughput fashion across many (20–50) samples and operating under compliant-ready conditions.
According to Figure 1 flow chart, the results for the HCP Discovery Assay are useful for compiling an HCP peptide library containing peptide sequence, precursor m/z and charge state, as well as retention time. The HCP Monitoring Assay is designed for targeted monitoring of a subset of HCP peptides across a variety of mAb production schemes. The HCP Monitoring dataset of MIX-5 spiked peptides was used for the development of the monitoring assay, mimicking a dataset containing various levels of HCP derived peptides.
To simulate an HCP monitoring experiment, four protein digests (ADH, BSA, ENL and PHO) were spiked at four different concentration levels in four NIST mAb digests, while one protein digest (CLP-B) was spiked at constant level (120 ppm) in all four samples. The monitoring of several spiked protein digests with known concentrations was used for simulating the typical samples resulted from mAb purification that would contain different concentrations of HCPs following various purification protocols.
The LC-MS data was acquired in MSE mode using a 30 minute gradient on a BioAccord LC-MS Ssystem. The MIX-5 spiked NIST mAb digest dataset composed of 25 runs, including five5 replicates for each sample, was then processed using the UNIFI Application Accurate Mass Screening Workflow available in waters_connect. Data acquisition and processing on the waters_connect platform can be routinely accomplished within a GMP laboratory environment, fulfilling all industry requirements.11,12
A list of the 43 best responding peptides in ESI-MS, comprising 10 peptides from each proteins (ADH, BSA, ENL, and PHO), along with 3 peptides from the CLP-B protein digest standard, was automatically imported in the Component Table, a section of the data processing method designating those peptides for targeted quantification. The list contained only the most abundant precursor charge state for each peptide. Extracted mass chromatograms were generated for all 43 spiked peptides and the corresponding chromatographic peaks were automatically integrated and ranked according to their peak area. The most abundant peptides from each protein (based on their peak area counts) were selected for Hi3 protein quantification.13 Concentration of four spiked proteins (ADH, BSA, ENL, and PHO) was measured against the CLP-B protein digest which was spiked at constant level (120 ppm) in all samples.
Each of the four spiked proteins can be easily tracked down to their corresponding lowest spiked levels across all spiked samples as exemplified by panels A-E from Figure 5. In this figure, a trend plot is displayed for each spiked protein by monitoring a single peptide from each protein: peptide VVGLSTLPEIYEK (precursor 724.41, +2, panel 5A) from ADH, peptide LVNELTEFAK (precursor 582.32, +2, panel 5B) from BSA, peptide LPQVEGTGGDVQPSQDLVR (precursor 665.68, +3, panel 5C) from CLP B, peptide TAGIQIVADDLTVTNPK (precursor 585.99, +3, panel 5D) from ENL, and peptide VAAAFPGDVDR (precursor 559.29, +2, panel 5E) from PHO. The HCP Monitoring Assay is clearly able to find all the spiked proteins in the NIST mAb digest and it achieved a detection limit consistent with the HCP Discovery assay (5 ppm) in the case of ADH (see panel 5A).



Conclusion
- Two analytical scale LC-MS workflows have been developed for HCP analysis: 1) A Discovery HCP workflow to identify and quantify unknown HCPs during development; 2) A Monitoring HCP to quantify known HCPs typically in a GMP environment supporting manufacture and quality analysis of biotherapeutics.
- Data-independent LC-MSE acquisition on the Xevo G3 QTof instrument, identified and quantified two spiked reference proteins, as well as seven endogenous HCPs from the NIST mAb reference material with an HCP Discovery Assay LLOQ of 5 ppm.
- The HCP Monitoring Assay, The HCP Monitoring Assay quantified four spiked proteins (MIX-4) in the NIST mAb digest sample with the same LLOQ (5 ppm) using the BioAccord LC-MS System and the Accurate Mass Screening workflow from waters_connect.
- Both assays were performed on compliant-ready informatics, allowing their use in regulated environments, such as QC or Manufacturing, or when internal data integrity concerns are present.
- Both workflows described here use robust, 2.1 mm ID, analytical scale separations, avoiding the reproducibility and robustness concerns of workflows using nanoscale LC coupled to HRMS.
References
- Doneanu CE, Anderson M, Williams BJ, Lauber MA, Chakraborty A, Chen W. Enhanced Detection of Low-Abundance Host-Cell Protein Impurities in High-Purity Monoclonal Antibodies Down to 1 ppm Using ion Mobility Mass Spectrometry Coupled with Multidimensional Liquid Chromatography, Anal Chem, 2015, 87, 10283–10291.
- Weibin C, Doneanu CE, Lauber MA, Koza S, Prakash K, Stapels M, Fountain KJ. Improved Identification and Quantification of Host Cell Proteins (HCPs) in Biotherapeutics Using Liquid Chromatography-Mass Spectrometry, book chapter in Technologies for Therapeutic Monoclonal antibody characterization, Vol 3, ACS Symposium Series, 2015, 357–393.
- Molden R, Hu M, Yen ES, Saggase D, Reilly J, Mattila J, Qiu H, Chen G, Bak H, Li N. Host Cell Protein Profiling of Commercial Therapeutic Protein Drugs as a Benchmark for Monoclonal Antibody-based Therapeutic Protein Development, MABS, 2021, 13, e1955811.
- Ito T, Lutz H, Tan L, Wang B, Tan J, Patel M, Chen L, Tsunakawa Y, Park B, Banerjee S. Host Cell Proteins in Monoclonal Antibody Processing: Control, Detection, and Removal, Biotechnol Prog, 2024, DOI: 10.1002/btpr.3448.
- Huang L, Wang N, Mitchell CE, Brownlee T, Maple SR, De Felippis MR. A Novel Sample Preparation for Shotgun Proteomics Characterization of HCPs in Antibodies, Anal Chem, 2017, 89, 5436–5444.
- Chen IH, Xiao H, Daly T, Ning L. Improved Host Cell Protein Analysis in Monoclonal Antibody Products through Molecular Weight Cutoff Enrichment, Anal Chem, 2020, 92, 3751–3757.
- Wang Q, Slaney TR, Wu W, Ludwig R, Tao Li, Leone A. Enhancing Host-Cell Protein Detection in Protein Therapeutics using HILIC Enrichment and Proteomic Analysis, Anal Chem, 2020, 92, 10327–10335.
- Yang F, Li D, Kufer R, Cadang L, Zhang J, Dai L, Gua J, Wohlrab S, Grennwood-Goodwin M, Shen A, Duan D, Li H, Yuk IH. Versatile LC-MS-based Workflow with Robust 0.1 ppm Sensitivity for Identifying Residual HCPs in Biotherapeutic Products, Anal Chem, 2021, 93, 723–731.
- Doneanu C, Gomes A, Williams B, Yu YQ, Chen W. Identification of Host Cell Proteins (HCPs) in Monoclonal Antibodies at Sub-ppm Levels using the SYNAPT XS Mass Spectrometer, 2021, Waters application note 720007101.
- Pilely K, Johansen MR, Lund RR, Kofoed T, Jorgensen TK, Skriver L, Mortz E. Monitoring process-related impurities in biologics – host cell protein analysis, Anal & Bioanal Chem, 2022, 414, 747–758.
- Guo J, Kufer R, Li D, Wohlrab S, Greenwood-Goodwin M, Yang F. Technical advancement and practical considerations of LC-MS/MS-based methods for host cell protein identification and quantitation to support project development, MABS, 2023, 15, 2213365.
- Ippoliti S, Zampa N, Yu YQ, Lauber MA. Versatile and Rapid Digestion Protocols for Biopharmaceutical Characterization using RapiZyme Trypsin, 2023, Waters application note 720007840.
- Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition, Mol Cell Proteomics, 2006, 5, 144–156.
720008448EN, July 2024