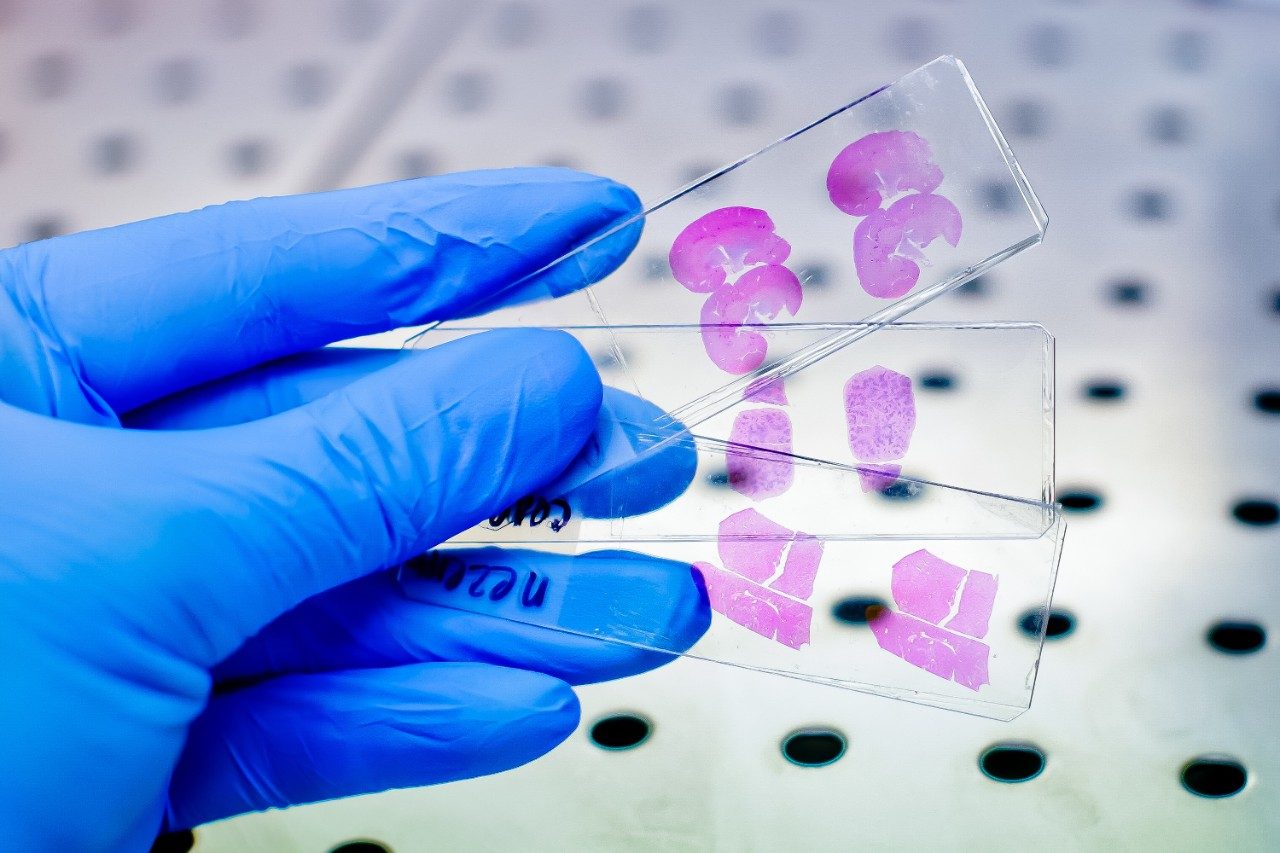
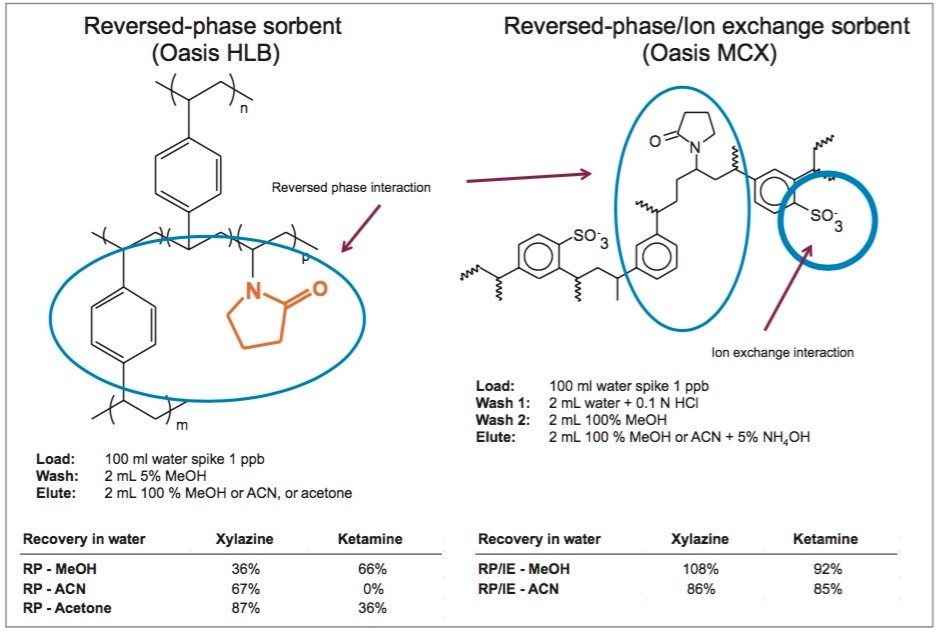
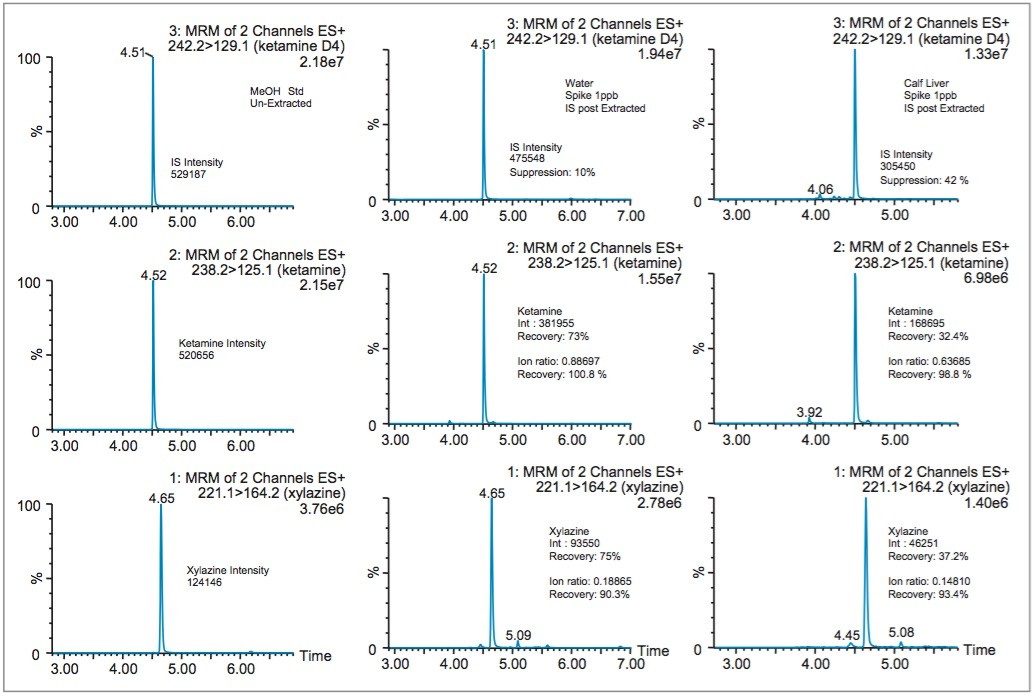
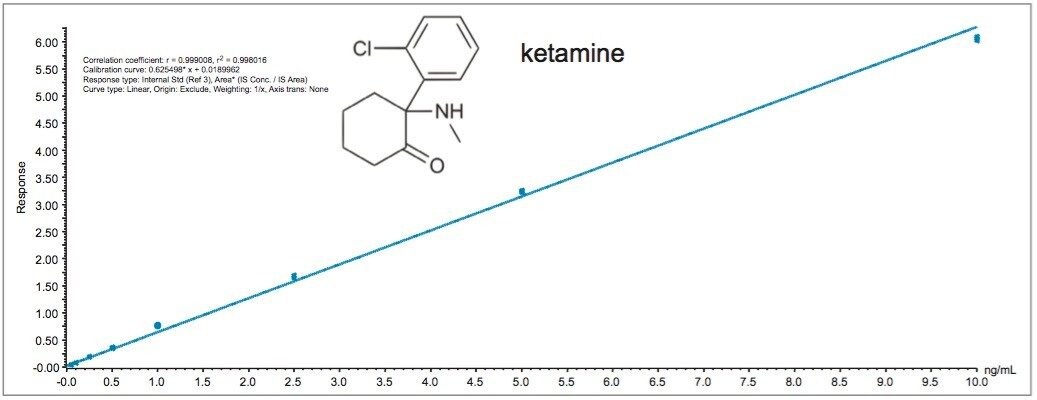
In most cases, acidic methanol or acetonitrile are the popular choices to extract a target analyte from a solid matrix. The homogenization process is typically performed with a common kitchen blender or using a hand-held homogenizer (ex: Polytron). Those techniques can be cumbersome and are difficult to apply to small mass samples. In recent years, novel developments with ceramic or stainless steel ball bearings in combination with high speed orbital shakers have shown the possibility to reach complete cell membrane breakdown in less than 60 seconds. With variable cycle speed, this novel homogenization protocol can process sample sizes from 0.1 to 5.0 grams. In this application, the mass range for the rat brain, heart, lung, liver, kidney, and spleen sample was between 0.2 and 0.8 grams. At this stage of the application, once a sample is completely homogenized, the sample is centrifuged, which will create a solid pellet on the bottom of the tube with the organic supernatant above. The organic supernatant is then decanted. Depending on the extraction conditions (pH and polarity), the target analyte is expected to be in solution and un-bound in the extraction solvent. In some applications, this crude extract can be used directly for quantification, however there is a high risk the raw sample extract will seriously reduce the robustness of the LC-MS/MS performance after a few injections.
In traditional SPE protocols, when the target analyte is dissolved in a high percentage of organic solvent, the supernatant is usually evaporated to dryness and reconstituted in an aqueous diluent for further cleanup. In instances where an evaporation to dryness step is needed, there is a risk of evaporative loss or possible re-dissolution issues. An effective way to avoid this lengthy step is to simply dilute the organic supernatant in a large aqueous volume at an organic/water ratio of less than 5%. A water volume between 100 and 200 mL is more than adequate to reach low organic ratio without any risk of breakthrough on the trapping column during loading phase. It may be perceived as a drawback, since the loading volume is quite large. However, with a loading flow rate at 10mL/min and using a large bore SPE barrel (6 cc with 150 mg bed mass), a 100 mL sample can be concentrated in 10 minutes, while evaporating to dryness can take several hours to complete.
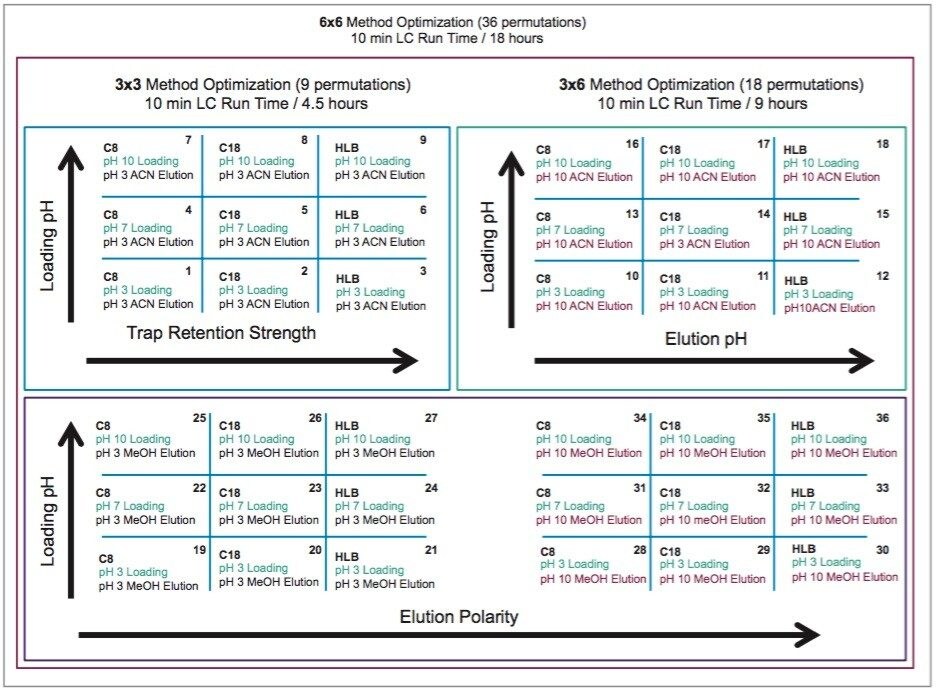
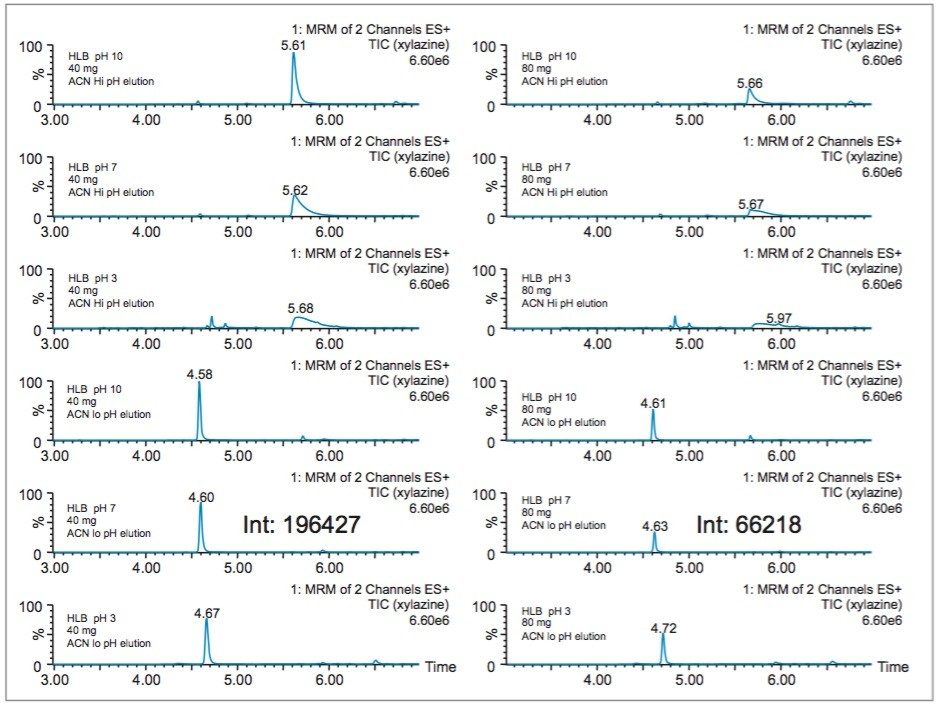
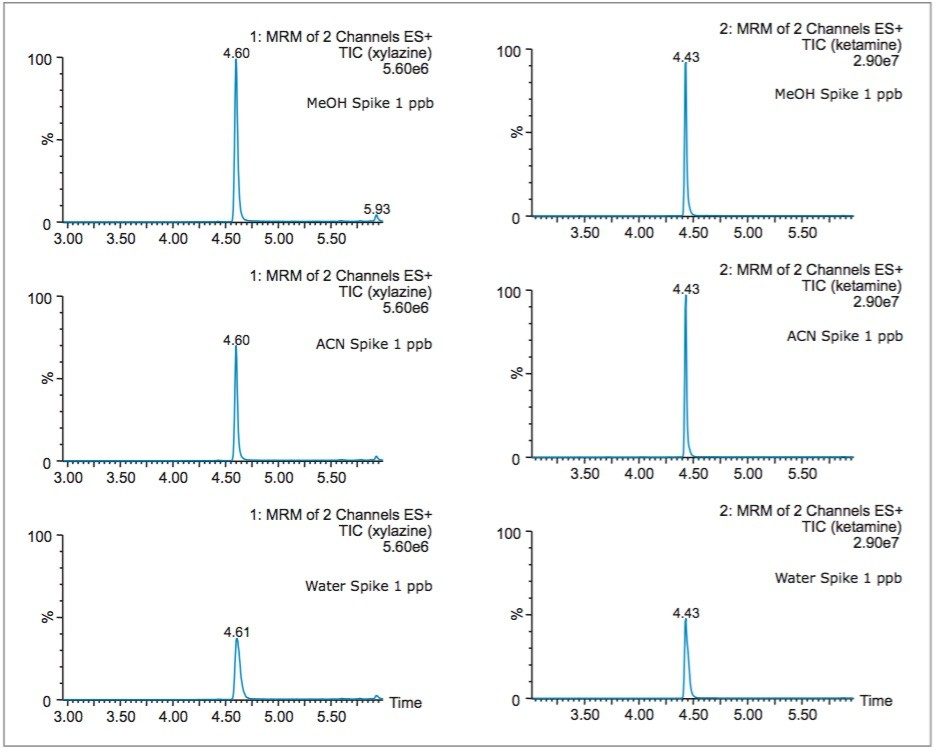
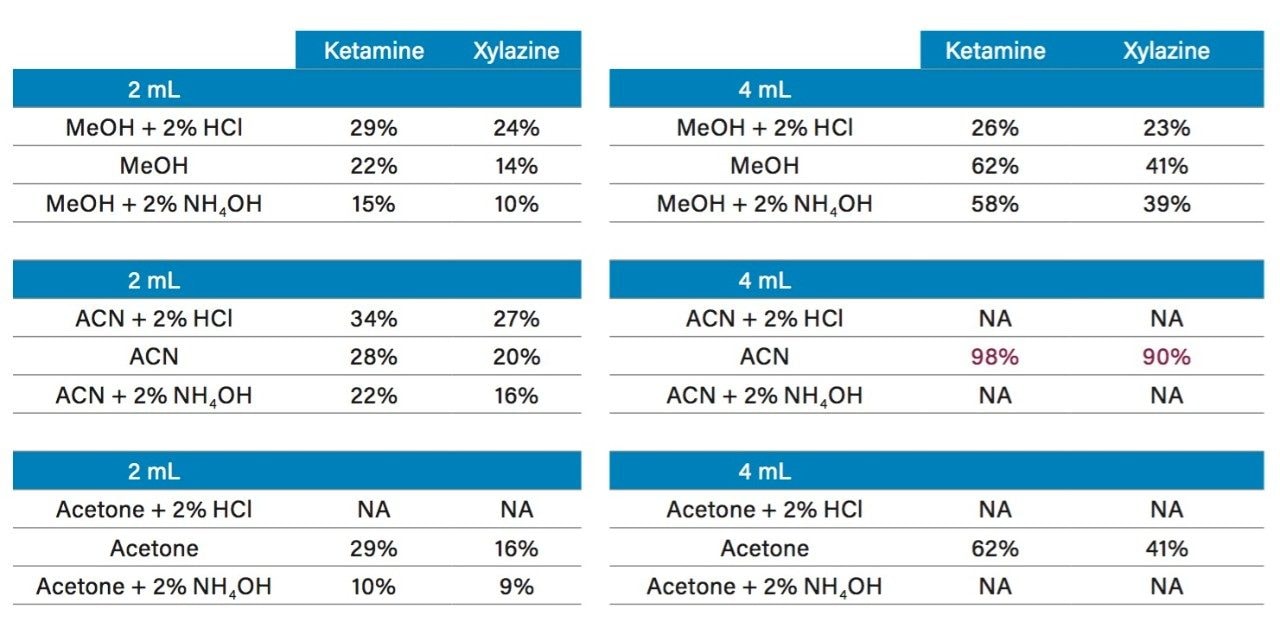
In this application, several organic solvents (MeOH, ACN, acetone), pH conditions (HCl vs NH4OH), and volume-to-mass ratio (2 mL vs 4 mL) were evaluated for extraction purposes while keeping the loading organic/water ratio below 5%. For this portion of the work, a 1 gram spiked calf liver sample was used for each optimization parameter. The results revealed one extraction condition in particular gave excellent recoveries (see Table 2), while other extraction conditions gave recovery values below 50%. In some samples, the solid/liquid extraction produced highly complex extracts, which subsequently created a rapid blockage during loading phase. Those extraction conditions were reported as non-available (NA) in Table 2. In keeping with an extraction time objective of 30 minutes or less, those conditions were not pursued further, however additional cleanup prior to the SPE loading could be beneficial (dispersive SPE, filtration, high speed centrifugation, etc.) thus resolving any potential clogging issues.