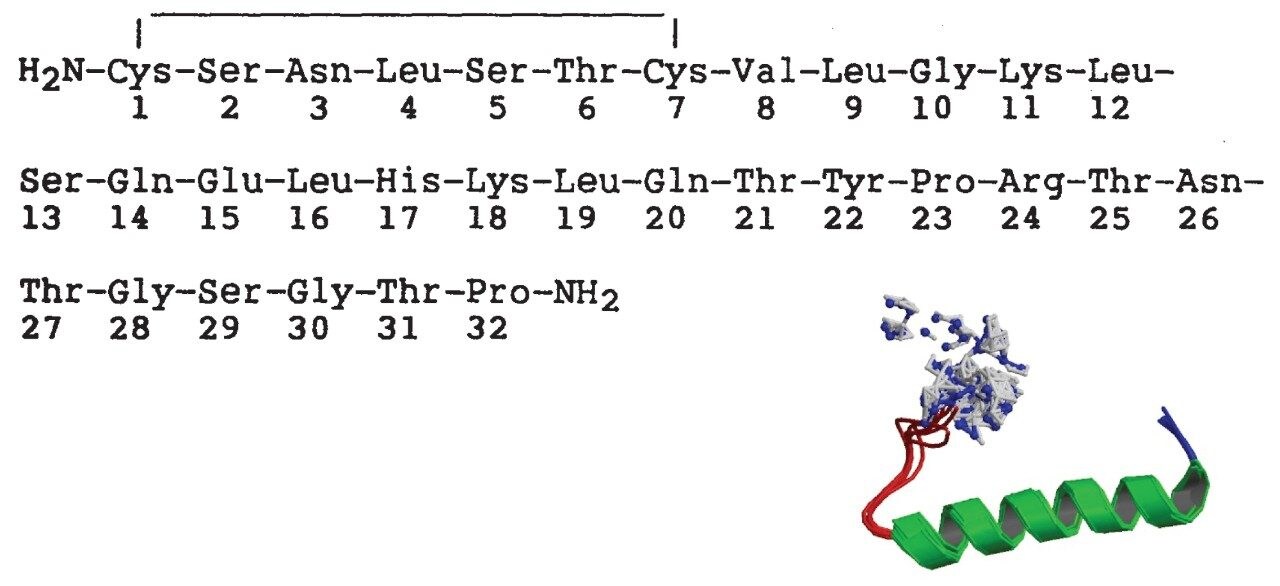
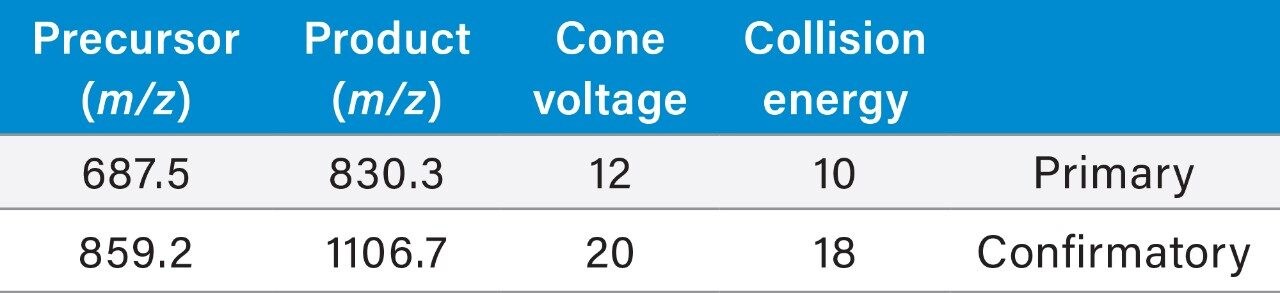
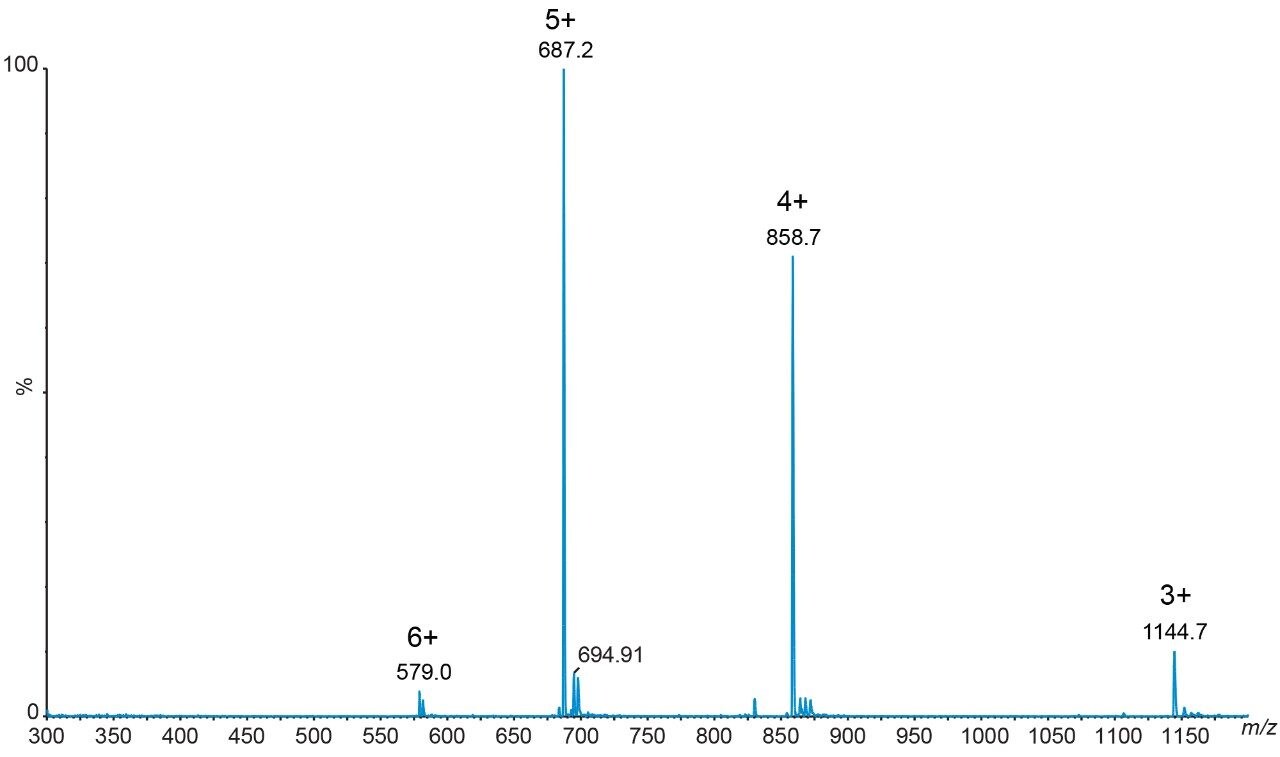
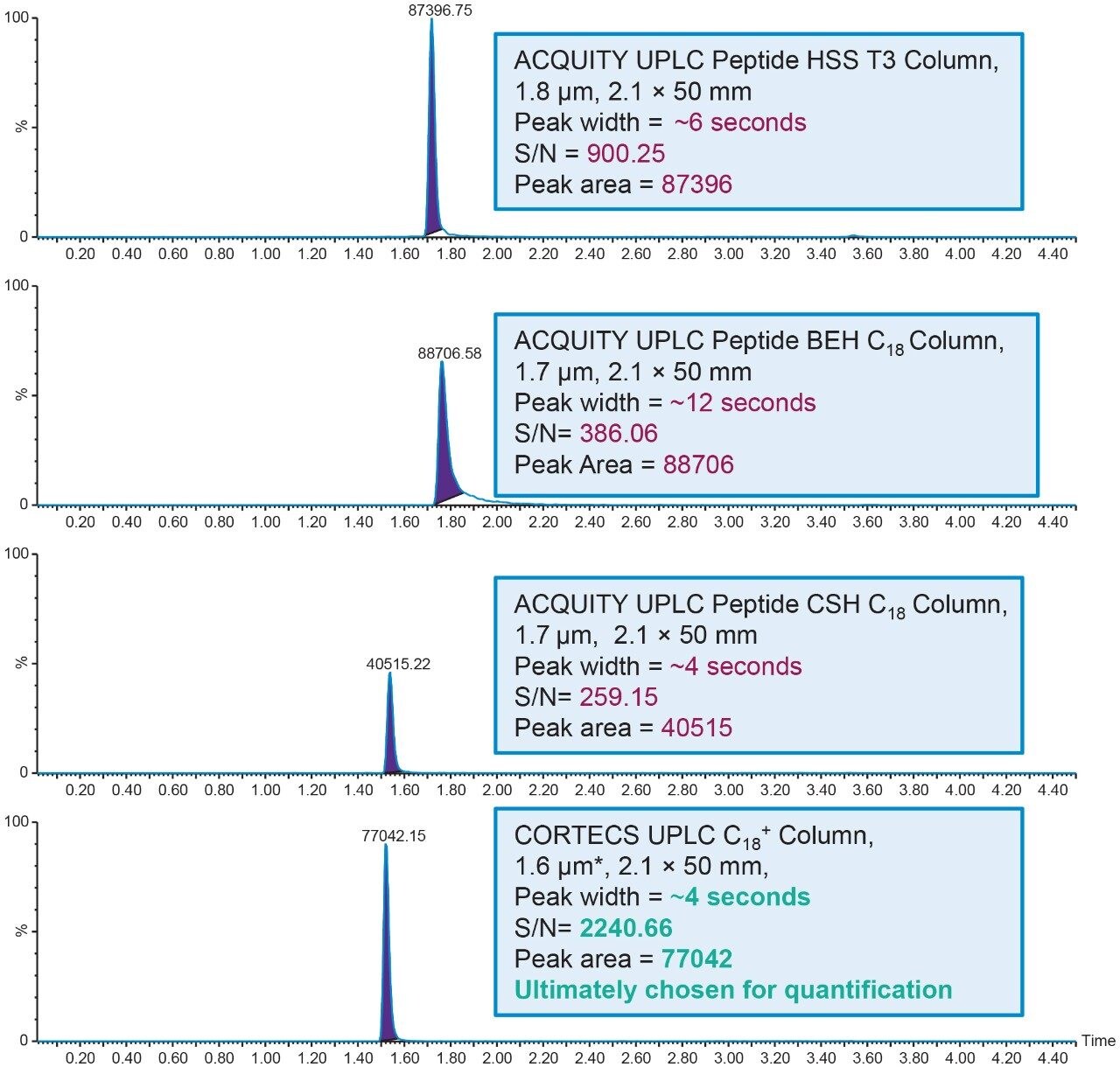
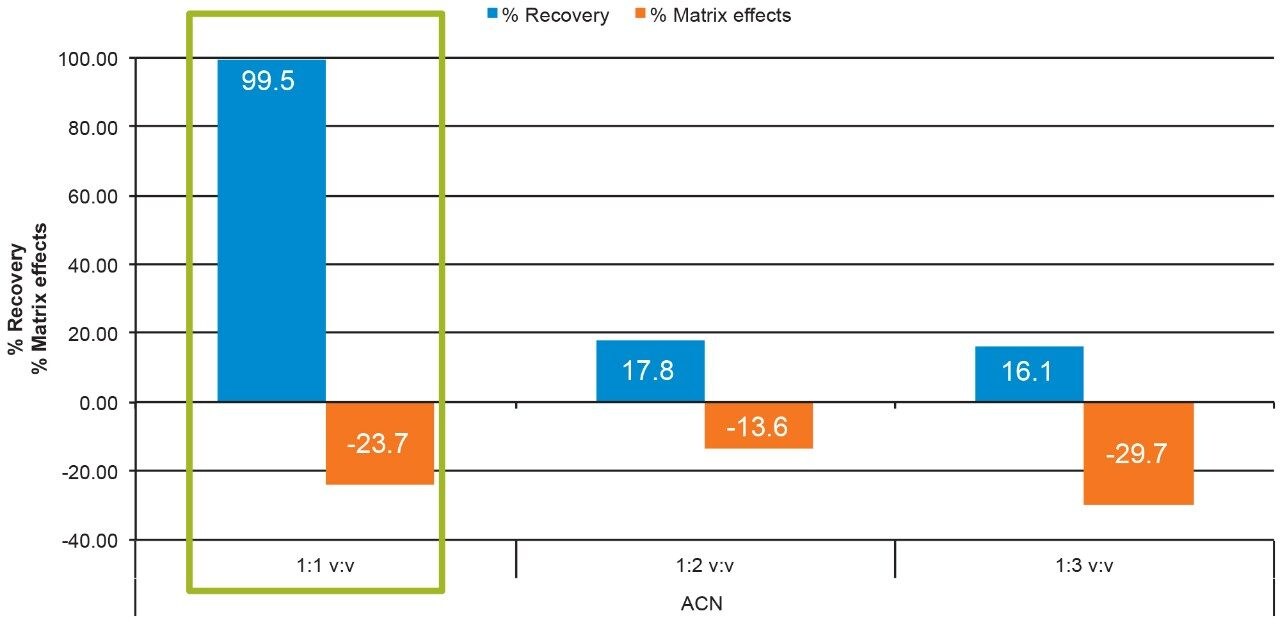
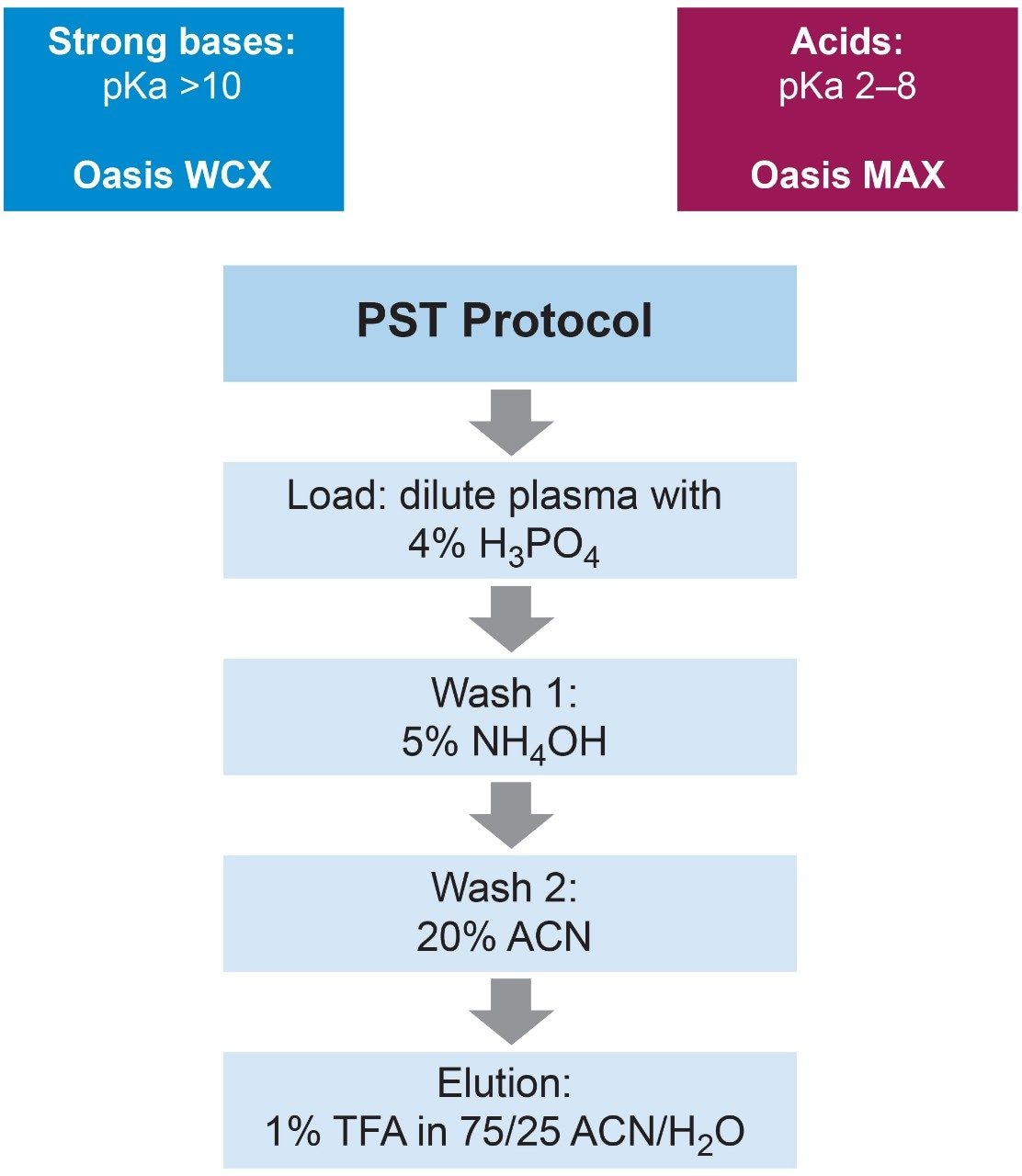
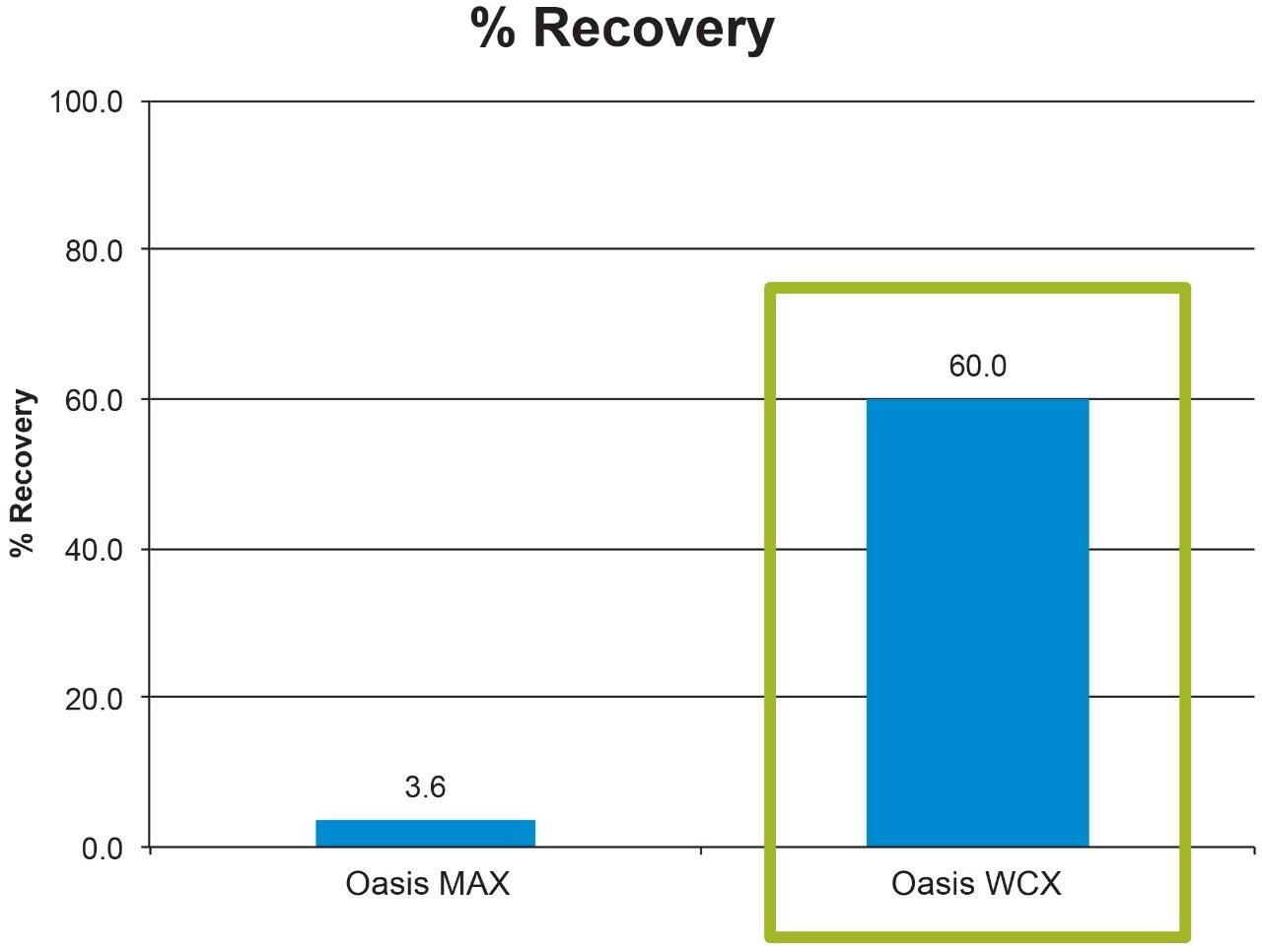
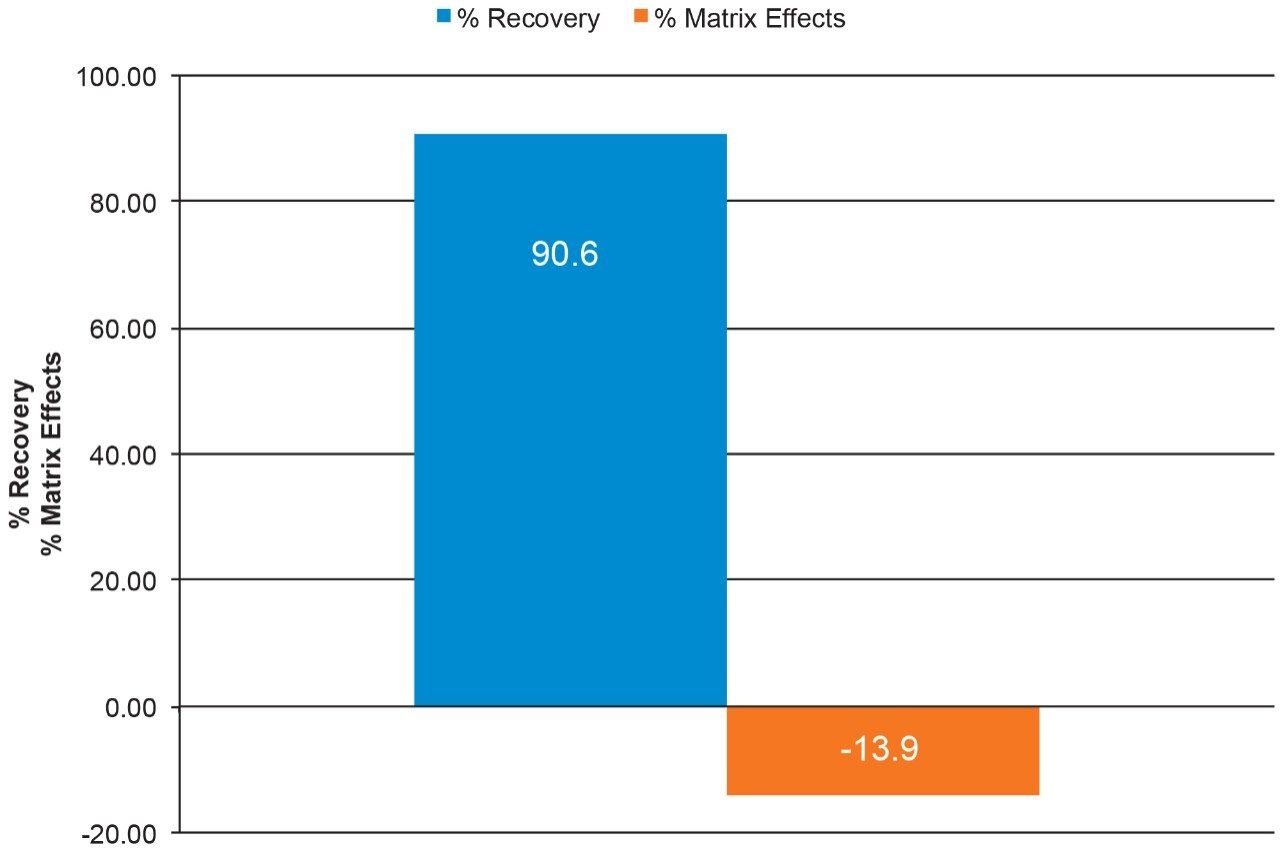
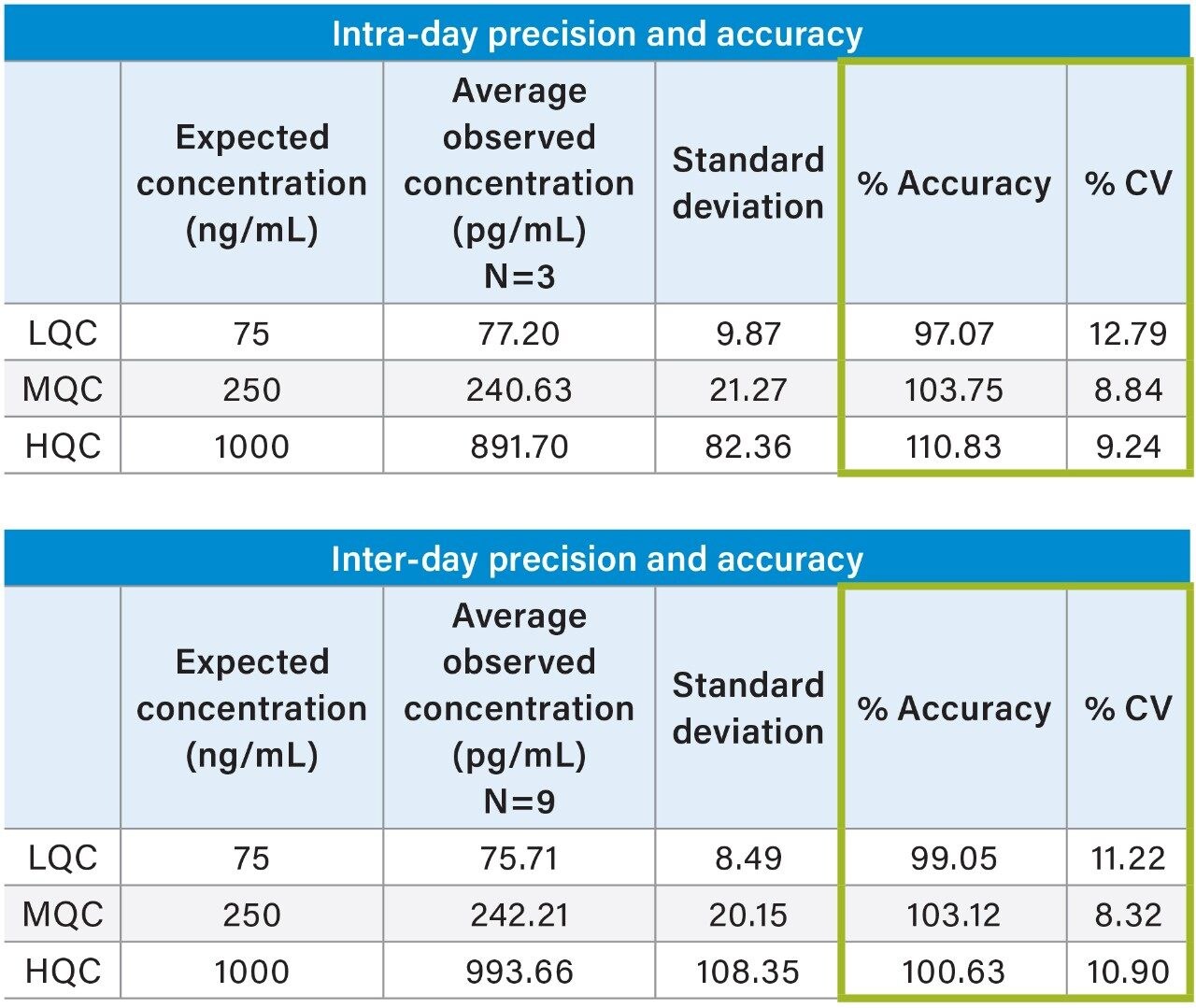
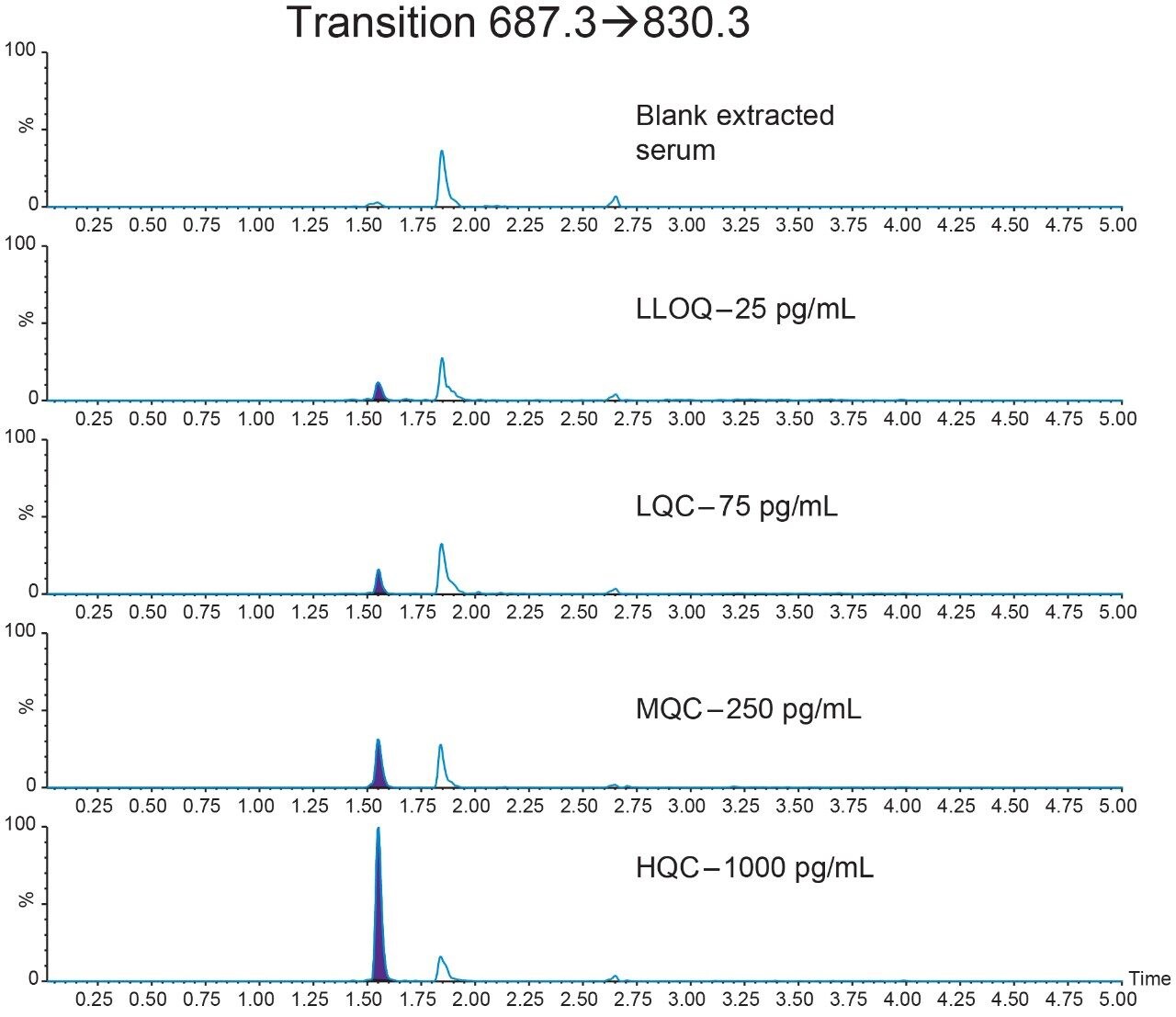
Salmon calcitonin (sCT) is a 32 amino acid (MW 3439.1 Da) synthetic polypeptide and is used in the treatment of Paget’s disease, osteoporosis, and hypercalcemia.1 Its amino acid sequence and structure are highlighted in Figure 1.2 Accurate quantification of sCT can be challenging, as the pharmacokinetics of sCT is characterized by rapid absorption within 30 minutes and rapid elimination with a half-life of ~1 hour, resulting in very low circulating plasma levels (pg/mL).2,3 Although peptide biologics like sCT have historically been quantified using ligand binding assays (LBAs), over the past few years, there has been a growing trend towards the bioanalysis of large molecules by LC-MS. This is, in part, driven by the fact that LBAs can suffer from cross-reactivity, lack of specificity, limited dynamic range, and long method development times. In contrast, LC-MS has the advantage of shorter development times, greater accuracy and precision, the ability to multiplex, and can readily distinguish between closely related analogues, metabolites, or endogenous interferences. Previously described LC-MS methods for salmon calcitonin have achieved a lower limit of quantification (LLOQ) of 50 pg/mL by tuning the tandem (triple) quadrupole to achieve a resolution of 0.2 Da as opposed to the unit resolution at which these instruments usually operate,3,4 while other methods have used larger sample volumes (500 μL) to achieve similar LLOQ’s. The work described here uses UPLC separation, tandem (triple) quadrupole MS and simple, fast, and selective sample preparation in a 96-well format to achieve an LLOQ of 25 pg/mL, extracted from 100 μL of serum.