Biotherapeutic Protein Characterization and Attribute Monitoring Using Xevo™ G3 QTof, BioAccord™ LC-MS System, and waters_connect™ Informatics
Abstract
As biosimilar monoclonal antibody (mAb) drug products become more prevalent, having efficient tools to properly assess their analytical comparability is highly desirable. These tools should enable detailed peptide level analysis, including localizing the post-translational modifications (PTMs) during peptide map characterization studies (attribute definition), and monitoring of the relative abundance of key product and process attributes across samples in development and commercialization. The compliant-ready waters_connect informatics platform provides efficient app-based automated workflows for carrying out both workflows from acquisition to analysis to reporting. In this study, characterization was performed using the peptide mapping workflow in the waters_connect UNIFI™ App on a Xevo G3 QTof mass spectrometer coupled with an ACQUITY™ Premier UPLC™ System. After peptide mapping data was processed, selected attribute peptides were added to a custom database within the Scientific Library and subsequently imported into the Peptide MAM App for targeted analysis. The Peptide MAM App was used to monitor the attributes across biosimilar samples and stress conditions on a BioAccord LC-ToF mass detection System. The results showcase the streamlined integrated workflows for attribute characterization and monitoring that spans multiple fit-for-purpose LC-HRMS instrument platforms.
Benefits
- High confidence peptide mapping of innovator and biosimilar mAbs with localization of modifications using the Xevo G3 QTof
- Seamless transition of methods across instruments within the network capable waters_connect Informatics Platform
- Efficient, compliance-ready workflow for MAM analysis of peptides on BioAccord System that supports targeted attribute monitoring and “new peak” detection-based purity assessments
Introduction
Before a monoclonal antibody (mAb) drug product can go to market, careful assessment of its critical quality attributes (CQAs) is necessary to ensure the manufacturing process can maintain the product’s safety and efficacy. This process includes characterization of the protein using peptide mapping, requiring both high sequence coverage of the mAb and localization of any modifications to facilitate the identification of potential CQAs, and then monitoring these attributes across samples as development and commercialization progresses. Changes in the abundance of CQAs provides insight into factors related to product efficacy, safety, and process robustness. Proper assessment of CQAs is important for new products coming to market, as well as for biosimilar drug products that have the same sequence but may have altered levels of some attributes due to manufacturing process differences. Biosimilarity determination involves both attribute comparisons of the biosimilar product to the innovator reference material and comparable profiles of stress and forced-degradation behaviors. Historically, this was a process requiring multiple orthogonal analytical techniques optimized for assessing individual attributes. However, the Multi-Attribute Method (MAM) is now increasingly employed for quantifying numerous targeted attributes over multiple samples using a single LC-MS analysis method.
While peptide maps for protein characterization typically require a high-resolution mass spectrometer with MSMS capability to ensure accurate identification of attributes, monitoring of said attributes can be performed on a simpler mass analyzer that is smaller in size, lower in cost, and possessing an ease of operation suited for non-MS experts. The integrated waters_connect Informatics platform enables a seamless transition of methods between LC-MS instruments in a compliance-ready environment (Figure 1). This study demonstrates how a peptide mapping and monitoring workflow can be carried out within the waters_connect informatics platform on the latest generation Xevo G3 QTof mass detector. The peptide maps of four infliximab samples, innovator Remicade™ and biosimilars Inflectra™, Avsola™, and Renflexis™ were analyzed for attributes including deamidation, oxidation, lysine clipping, and glycosylation. Peptide characterization on the trypsin-digested mAb samples was performed on a Xevo G3 QTof instrument for identification and localization of post-translational modifications (PTMs) using the integrated UNIFI Application in waters_connect. Monitoring of attributes was performed with a BioAccord LC-MS System using the integrated Peptide MAM Application.2 Attributes were reproducibly monitored across biosimilars and across time points in a thermal stress study.

Experimental
Sample Description
Infliximab samples were thermally stressed for zero (no stress), one, or two weeks at 37 °C, then reduced, alkylated, desalted, tryptic digested, acidified to 0.1% formic acid, and diluted to a final concentration of 0.2 µg/µL.
MS Conditions
|
LC System: |
ACQUITY Premier UPLC System – BSM Configuration |
|
Detection: |
ACQUITY Premier TUV; 10 mm analytical flow cell; λ=214 nm |
|
Vials: |
QuanRecovery™ with MaxPeak HPS vials (p/n: 186009186) |
|
Column(s): |
ACQUITY Premier Peptide CSH C18 130 Å 1.7 µm, 2.1 x 100 mm |
|
Column temperature.: |
60 °C |
|
Sample temperature.: |
8 °C |
|
Injection volume: |
2 μL |
|
Flow rate: |
0.200 mL/min |
|
Mobile phase A: |
0.1% formic acid in water (LC-MS grade) |
|
Mobile phase B: |
0.1% formic acid in acetonitrile (LC-MS grade) |
|
Gradient: |
1–35 %B over 50 min gradient (80 min total run time) |
MS Conditions for Characterization
|
MS system: |
Xevo G3 QTof |
|
Ionization mode: |
ESI+ |
|
Acquisition range: |
100–2000 m/z |
|
Capillary voltage: |
2.2 kV |
|
Collision energy: |
Low energy: 6 V High energy ramp: 20–50 V |
|
Cone voltage: |
20 V |
|
Source temperature.: |
120 °C |
|
Desolvation temperature.: |
350 °C |
|
Cone gas: |
35 L/hr |
|
Desolvation gas: |
600 L/hr |
|
Intelligent Data Capture (IDC): |
Low (5) |
MS Conditions for Monitoring
|
MS system: |
BioAccord System |
|
Ionization mode: |
ESI+ |
|
Acquisition range: |
50–2000 m/z |
|
Capillary voltage: |
1.2 kV |
|
Cone voltage: |
30 V |
|
Fragmentation cone voltage: |
60–120 V |
|
Desolvation temperature: |
350 °C |
|
Intelligent Data Capture (IDC): |
On |
Data Management
Data were acquired and processed using the waters_connect informatics platform (version 2.2.0) with the integrated UNIFI Application (version 3.1.0.16) and Peptide MAM Application (version 1.5.0.13).
Results and Discussion
Before a biosimilar mAb drug product can be released, CQAs must be identified and relatively quantified compared to the originator product. Previously, we have shown how the workflow can be completed on a Xevo G3 QTof mass spectrometer.3 Here, we demonstrate how the workflow can be moved to a QC-friendly BioAccord LC-MS System for monitoring of attributes following characterization on the Xevo G3 QTof (Figure 1).
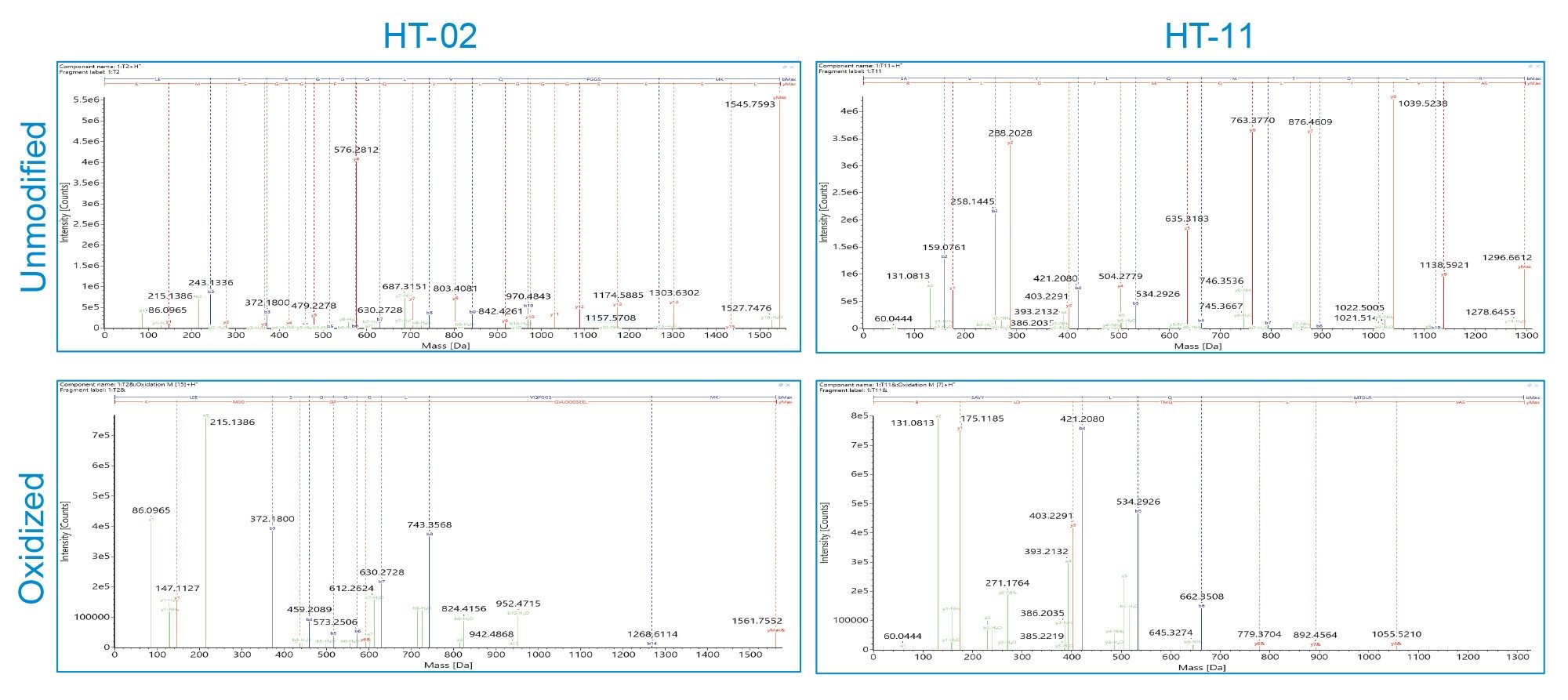
Trypsin-digested samples of each mAb were analyzed with an ACQUITY Premier UPLC System coupled to a Xevo G3 QTof mass spectrometer. MSE (Data Independent Acquisition) was used to identify peptides, including low level modified peptides. Data were acquired using the waters_connect UNIFI App and processed using the integrated peptide mapping workflow. Each infliximab sample was characterized with greater than 95% sequence coverage based on accurate mass (within 5 ppm) and fragment ions in the high energy data channel (b/y ions ≥3). Additionally, six oxidation, twelve deamidation, and 28 glycosylation PTMs were identified. Figure 2 shows high energy MSE spectra of two peptides, heavy chain tryptic peptide (HT)-02 and HT-11, in the unmodified and oxidized forms. The high coverage of matching fragment ions across each peptide’s amino acid backbone enables both confident identification of peptide assignments and localization of modifications to discrete amino acids within the peptide sequence. As the spectra were acquired with data independent acquisition, method development was simplified while still ensuring high-quality assignments.
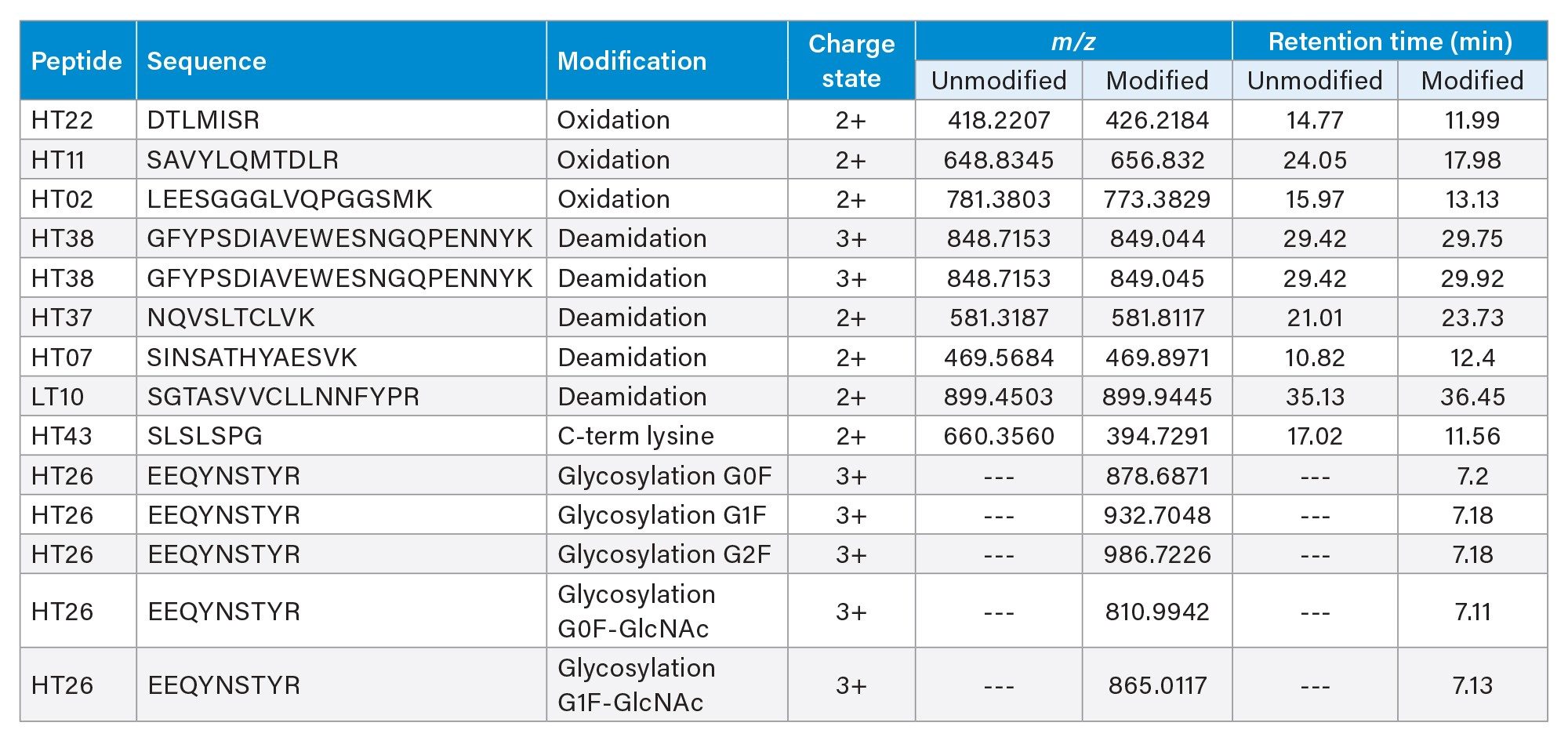
To establish analytical comparability for each biosimilar, attributes can be selected for monitoring in a MAM workflow within the waters_connect Informatics platform, which includes streamlined data acquisition, processing, and reporting. Each peptide selected for targeted monitoring, including modified and unmodified forms, can be added to a custom Scientific Library. The scientific libraries in waters_connect serve as repositories for components that can be later used for targeted analysis, storing information related to mass, charge, and retention time, among numerous other properties. Scientific libraries can be created for each molecule without the need to manually enter data and while maintaining data traceability. Upon selection of attributes to be monitored, they can be imported from the scientific library into the analysis method in the Peptide MAM App for monitoring. Table 1 shows the list of attributes selected for monitoring in this study.
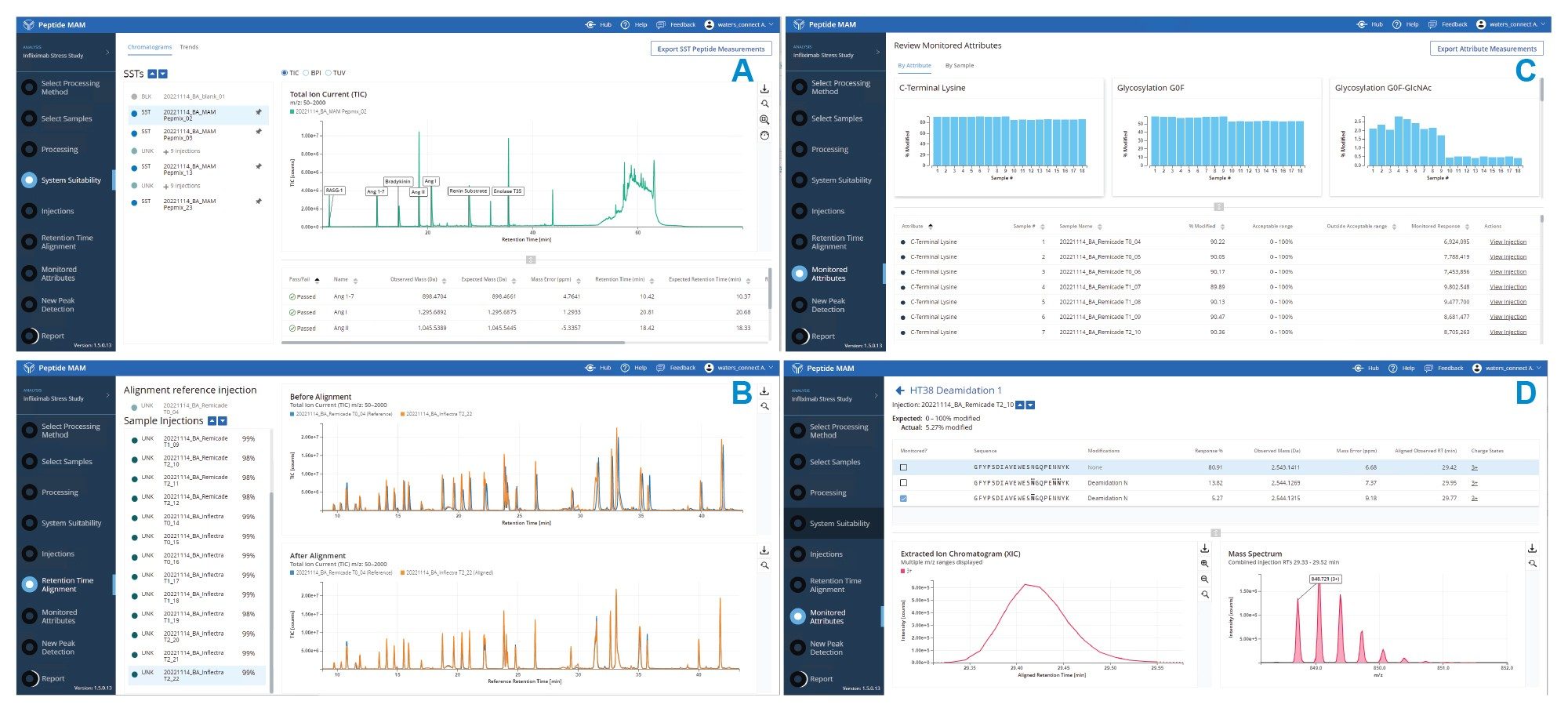
Monitoring of attributes across various sample sets was executed with a BioAccord LC-MS system, a fit-for-purpose TOF based HRMS platform with high usability that generates robust and high-quality results for attribute-based analysis. Here, we used the subset of attributes identified on the Xevo G3 QTof MS. The attributes were monitored across samples of infliximab biosimilar products and samples exposed to forced degradation in a stress study. System suitability injections were intercalated with sample injections to ensure the system was producing results within established performance specifications. MassPREP™ Peptide Mixture (p/n: 186002337) was used for these system suitability injections. These injections are flagged in the Peptide MAM App’s sample list and are evaluated based on specified criteria, including mass error, retention time error, response, and peak width. After processing results in the Peptide MAM App, the system suitability injections can be reviewed separately, as shown in Figure 3A. The peptide measurements can be visualized in each chromatogram or as trends across all system suitability injections, enabling both assessment of pass/fail criteria for each injection and identification of systematic shifts over the course of a sample sequence.
When monitoring attributes across a sample sequence, retention time alignment is performed on all sample injections against a designated reference injection to ensure appropriate peak codetection. An example of retention time alignment is shown in Figure 3B for an injection at the end of a long sequence. The top overlaid chromatograms (blue for reference and orange for current) show before alignment, and the bottom show after alignment. While the chromatograms are closely matched before alignment due to the robust performance of the UPLC system, retention time alignment enables near-perfect overlap of chromatograms from all injections. After reviewing the retention time alignment results, the relative quantification of monitored attributes can then be readily viewed as bar graphs either by sample or by attribute, as shown in Figure 3C, enabling rapid evaluation of the data. Individual attributes can be closely inspected in the data by clicking the “View Injection” tab, which brings up the information shown in Figure 3D. Here, peaks corresponding to each peptide form can be viewed in the chromatogram, as well as in the mass spectrum.
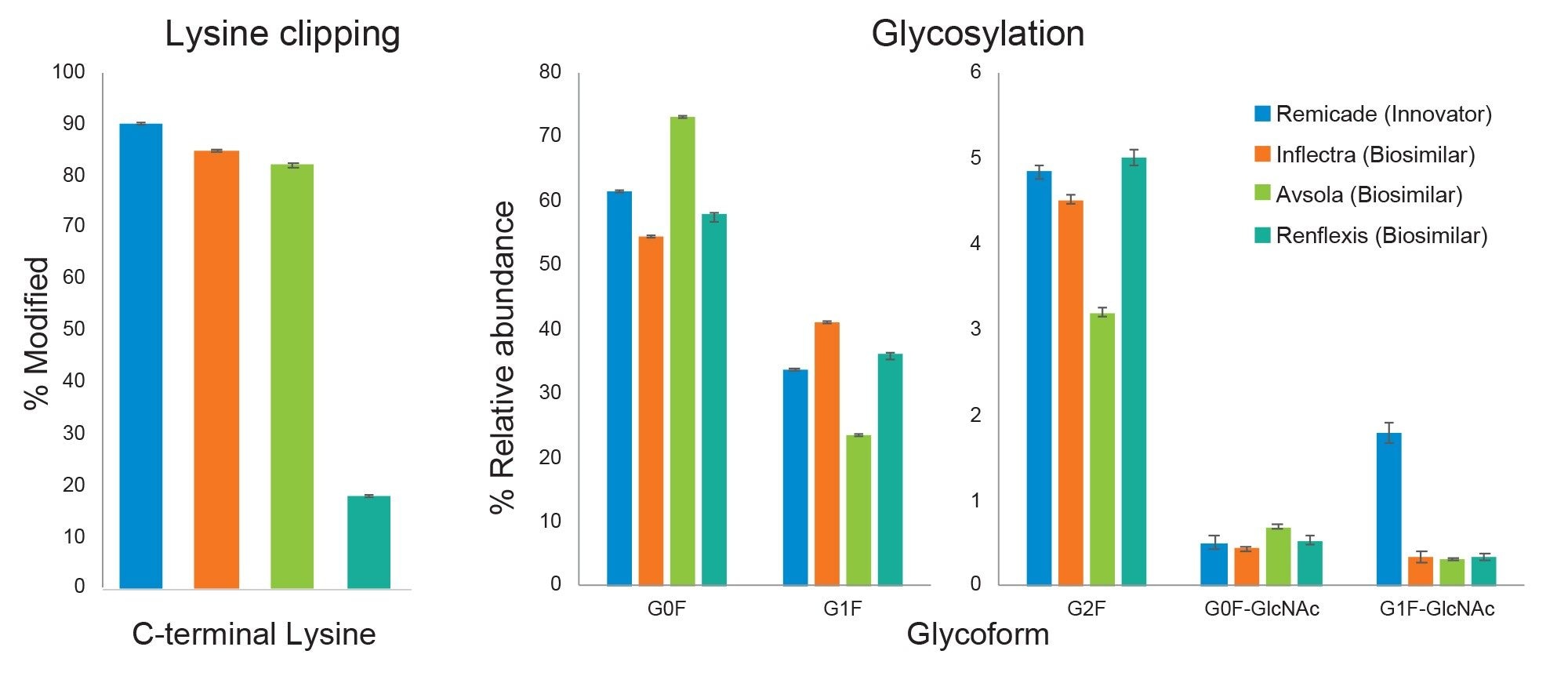
The Peptide MAM App was used to generate relative quantification results for C-terminal lysine clipping and glycosylation across the four infliximab products. Figure 4 summarizes the results comparing the three biosimilars, Inflectra, Avsola, and Renflexis, to the innovator, Remicade. An average relative standard deviation below 4% was observed for triplicate injections of each sample. This reproducibility enabled differences to be determined in the relative levels of peptide forms monitored. The abundance of C-terminal lysine was similar for Remicade, Inflectra, and Avsola, at 80-90%, while this value was much lower for Renflexis, at less than 20%. This measurement is important, as lysine clipping is a common modification during bioproduction and may impact receptor-binding.4
The abundances of various N-glycoforms of the HT-26 peptide were also different between the samples. Differences in glycosylation are expected, as the products originate from different cell lines (Remicade and Inflectra from murine and Avsola and Renflexis from Chinese Hamster Ovary cell line), but quantifying these differences is important because they may impact the immunogenicity, PK, and receptor interactions of the drug product. While G0F was the most prominent glycoform in all four samples, it was in the greatest abundance in Avsola at 73% and the lowest abundance in Inflectra at 54%. The second most prominent glycoform, G1F, varied from approximately 20 to 40%. The less-prominent glycoforms, G2F, G0F-GlcNAc, and G1F-GlcNAc, also varied across samples. Most notably, G1F-GlcNAc varied from ~2% in Remicade to ~0.3% in the three biosimilars.
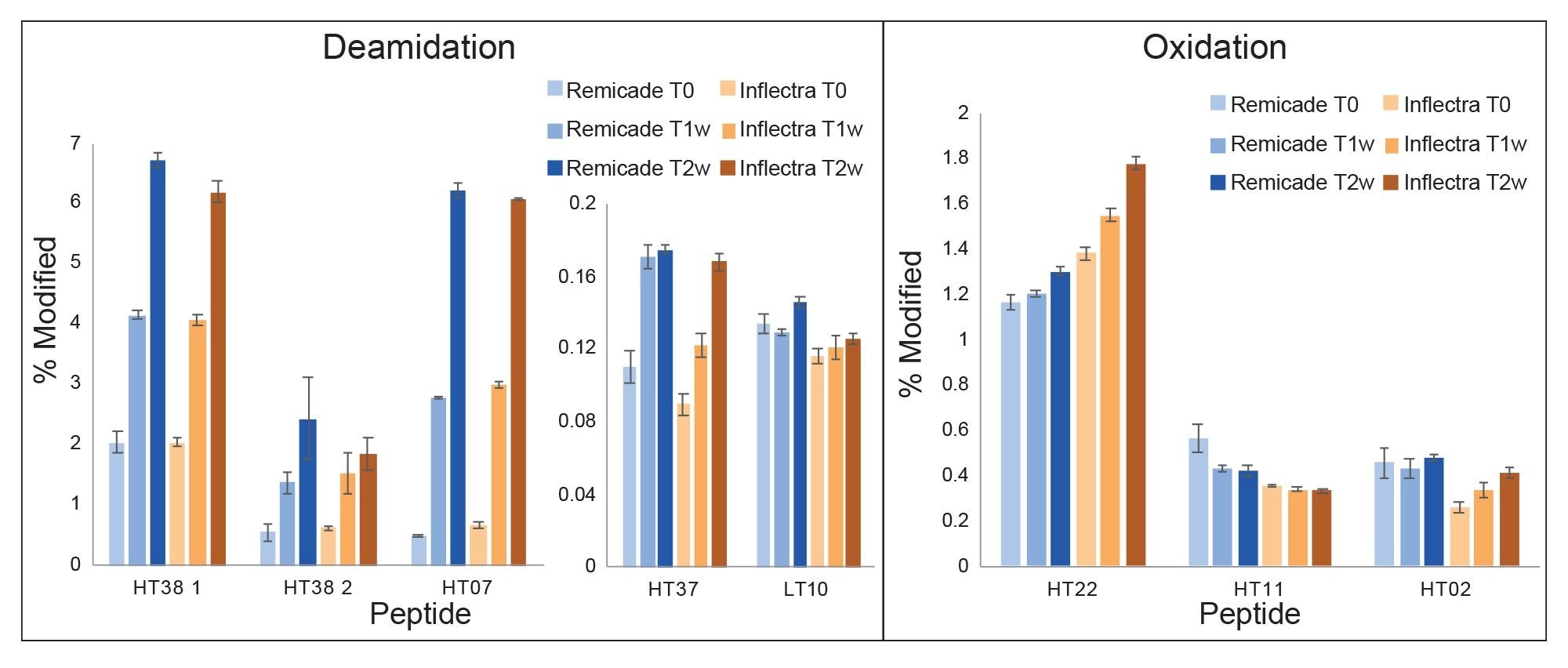
To further investigate sample differences, the innovator and one biosimilar, Inflectra, were subjected to a forced degradation stress study. Accelerated stress studies are helpful in evaluating the shelf life and stability of drugs by subjecting them to conditions at or above the upper limit of what they would be expected to encounter.5 Here, samples were subjected to thermal stress at 37 °C for one and two weeks, and the extent to which several peptides were oxidized or deamidated was quantified. The results are summarized in Figure 5, where blue indicates Remicade, orange indicates Inflectra, and darker shades indicate longer durations of stress. Generally, the % deamidation for each peptide was in agreement between the two samples and increased with increasing duration of heat-induced stress. Peptide oxidation did not have a clear trend in either sample, though differences were observed. Biosimilars are not expected to exhibit identical responses to stress but are expected to degrade by similar pathways. However, any differences observed require risk assessment to determine potential impacts to the safety and appropriate handling of the drug product.
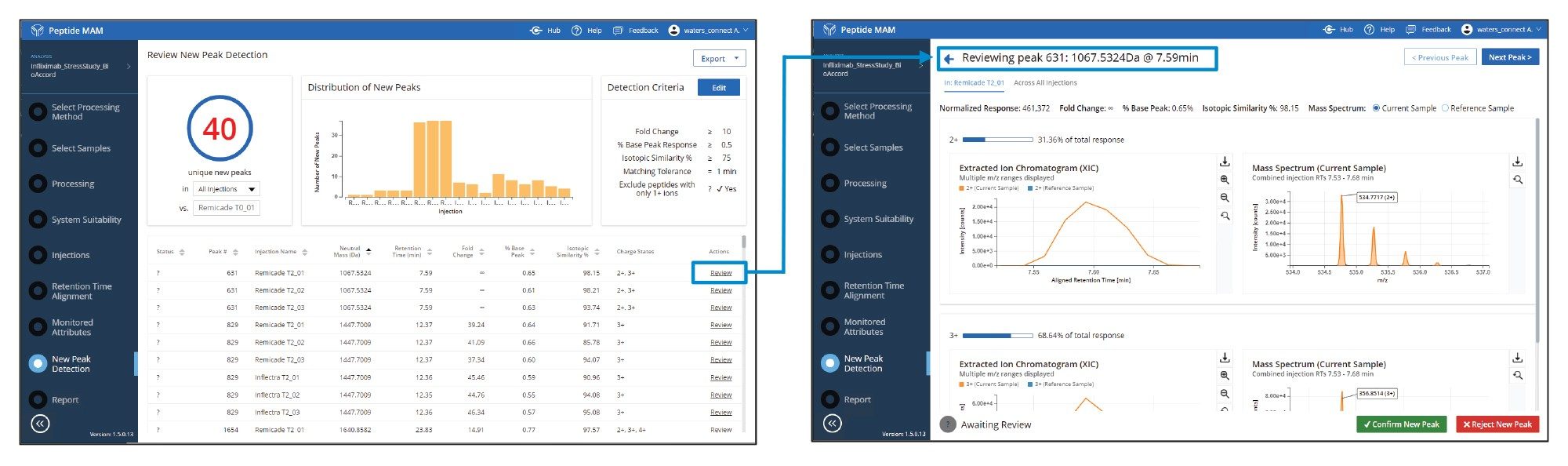
In addition to relative quantitation of key and critical attributes, it is often important to monitor for “new peaks” that have significant fold changes in the Biosimilar sample for compatibility assessment. New peak detection is a key element of peptide MAM workflows used as tests of product purity as they recognize new or changed features in the MS data channel, i.e. “peaks”, that are not present in the reference sample and not included in the list of targeted attributes. Details on how NPD (New Peak Detection) works in the MAM app was published in a recent application note.6 This step is useful for impurity analyses or for rapidly assessing differences in stressed samples. The New Peak Detection feature detected 40 “New Peaks” in the stressed samples using the criteria shown in Figure 6. Characteristics of these peaks are summarized in the left display. Each peak can be further investigated by clicking the “Review” link, which opens the data review window for new peaks. Here, each peak can be rejected if it is determined by the user as a false positive or marked confirmed if it is a new peak warranting further investigation. Confirmed new peaks can then be quickly identified from the same data set with the UNIFI App Peptide Mapping workflow if the fragmentation data from the MSE high energy data channel shows enough characteristic b/y ions for assignment confirmation, or through subsequent investigations using targeted peptide MSMS. The example shown in Figure 6 was identified in peptide mapping data processing to be a missed cleavage of the HT 9-10 peptide, which is likely an artifact of the sample preparation, not a stress-related impurity. Regardless, any new peaks can then be flagged for further investigation as necessary.
Conclusion
While biosimilar mAbs have an identical primary sequence to the innovator product, differences in production often lead to variation in attribute profiles between drug products. In evaluating biosimilarity, it must be established that these differences observed during analytical comparability studies would not be clinically relevant. Consequently, characterization and monitoring of product attributes is an important step in bringing a biosimilar drug product to market. Here, we demonstrated how the waters_connect Informatics platform facilitated this compliance-ready MAM workflow, even across different instrument systems operated under the waters_connect informatics platform. The Xevo G3 QTof mass spectrometer enabled high sequence coverage and localization of PTMs, which were then selected from the results table for potential CQA monitoring. This list can then populate a peptide MAM monitoring method for relative targeted quantification and new peak detection across large sample sets. This workflow was deployed for both a comparison of biosimilar products to the innovator reference and a stress study with these molecules. The seamless integration of the two peptide analysis workflows through the custom scientific library capability of the waters_connect platform is an effective way of transferring knowledge from characterization to monitoring functions that can be deployed across the responsible analytical laboratory organizations.
References
- Rogers RS, Nightlinger NS, Livingston B, Campbell P, Bailey R, Balland A. Development of a Quantitative Mass Spectrometry Multi-Attribute Method for Characterization, Quality Control Testing, and Disposition of Biologics. mAbs. 2015 Aug, 7(5), 881-890.
- Ranbaduge N, Yu YQ. A Streamlined Compliant Ready Workflow for Peptide-Based Multi-Attribute Method (MAM). Waters Application Note. 720007094, December 2020.
- DeLaney K, Ippoliti S, Reid L, Cornwell O, Yu YQ, Harry E, Towers M. Applying Peptide Mapping and Multi-Attribute Method (MAM) Workflow for Biosimilar mAb Drug Products Comparison on the Xevo G3 QTof Platform. Waters Application Note, 720007632, May 2022.
- Faid V, Leblanc Y, Berger M, Seifert A, Bihoreau N, Chevreux G. C-terminal Lysine Clipping of IgG1: Impact on Binding to Human FcγRIIIa and Neonatal Fc Receptors. Eur J Pharm Sci. 2021 Jan, 159, 105730.
- Pisupati K, Benet A, Yian Y, Okbazghi S, Kang J, Ford M, Saveliev S, Sen KI, Carlson E, Tolbert TJ, Ruotolo BT, Schwendeman SP, Schwendeman A. Biosimilarity Under Stress: A Forced Degradation Study of Remicade® and Remsima™. mAbs. 2017 Aug, 9(7), 1197-1209.
- Ranbaduge N, Yu YQ. Extending the Capabilities of the BioAccord LC-MS System with a Streamlined Workflow for Compliant-Ready Multi-Attribute Method (MAM). Waters Application Note, 720006963, July 2020.
Featured Products
720007914, May 2023