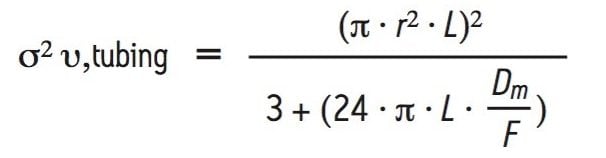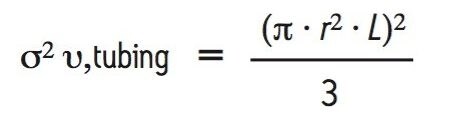Extra-column dispersion
Most of the chromatographic separation modes commonly used for the analysis of biotherapeutic proteins rely on a gradient elution of analytes that have been pre-concentrated at the head of the column (e. g. cation exchange and reversed phase). In this format, pre-column dispersion does not significantly degrade chromatographic performance. SEC, however, is operated in an isocratic mode and as a result, in addition to post-column dispersion that is observed for all of these modes, the performance of the SEC separation is far more prone to degradation due to pre-column dispersion. One of the primary advantages of using sub-2 µm particle columns for SEC analysis is the significant decrease in peak dispersion that these columns can provide relative to the larger 5–10 µm particle sizes typically used for SE-HPLC. However, this benefit can be easily obscured by the effects of extra-column dispersion. In this respect, the chromatographic instrumentation used and its configuration play key roles in the dispersion of the analyte peak(s).
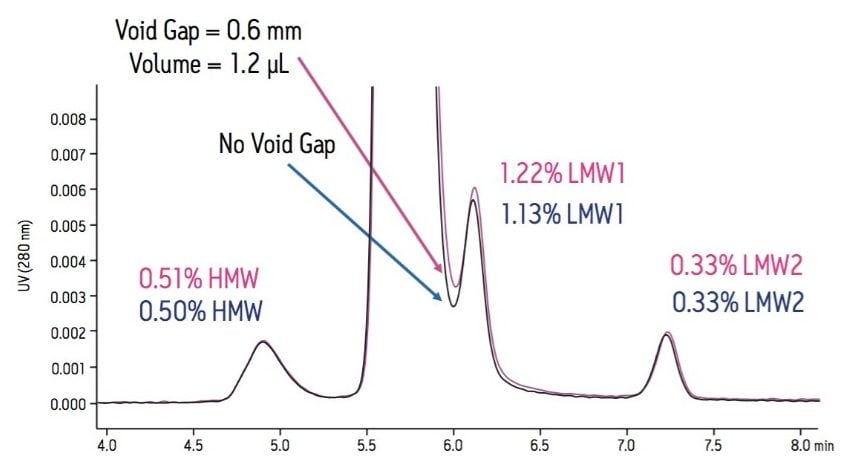
With respect to quantitative analysis, the effect of extra-column dispersion is most problematic in SE-UPLC separations in which there are partially resolved analytes. Separations where the peak area of the later eluting analyte of the critical pair is very low in comparison to that of the preceding peak are particularly sensitive to these effects. Once a low-dispersion chromatographic system has been selected and its performance optimized, selection of appropriate tubing and fittings must be made.
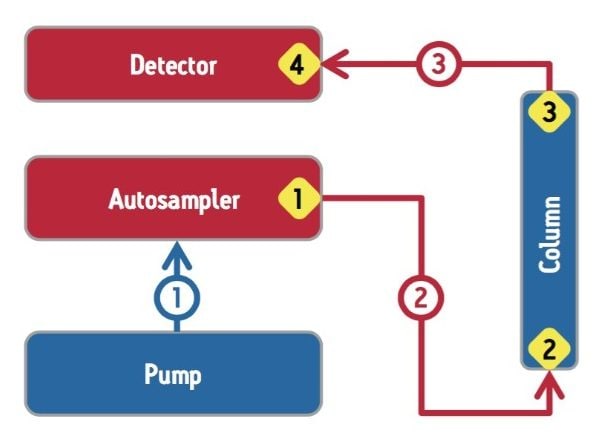
For a UV-absorbance based SE-UPLC method, the two system components that contribute to extra-column dispersion are the autosampler and the detector. Low dispersion systems specifically designed to operate at the high pressures needed to realize the full benefits of SE-UPLC are required for this methodology. In this regard, the performance of prospective instrumentation under the expected operating conditions should be evaluated thoroughly.
In addition to the UPLC system, significant extra-column dispersion can also be introduced by the tubing and fittings used to configure the SE-UPLC column into the chromatographic system. The extent of extra-column dispersion contributed by the capillary tubing used in a UPLC system may be estimated by a transition equation that was derived from the Taylor-Aris expression, which defines dispersion in long tubes and the equation defined by Attwood and Golay for dispersion in short tubes: