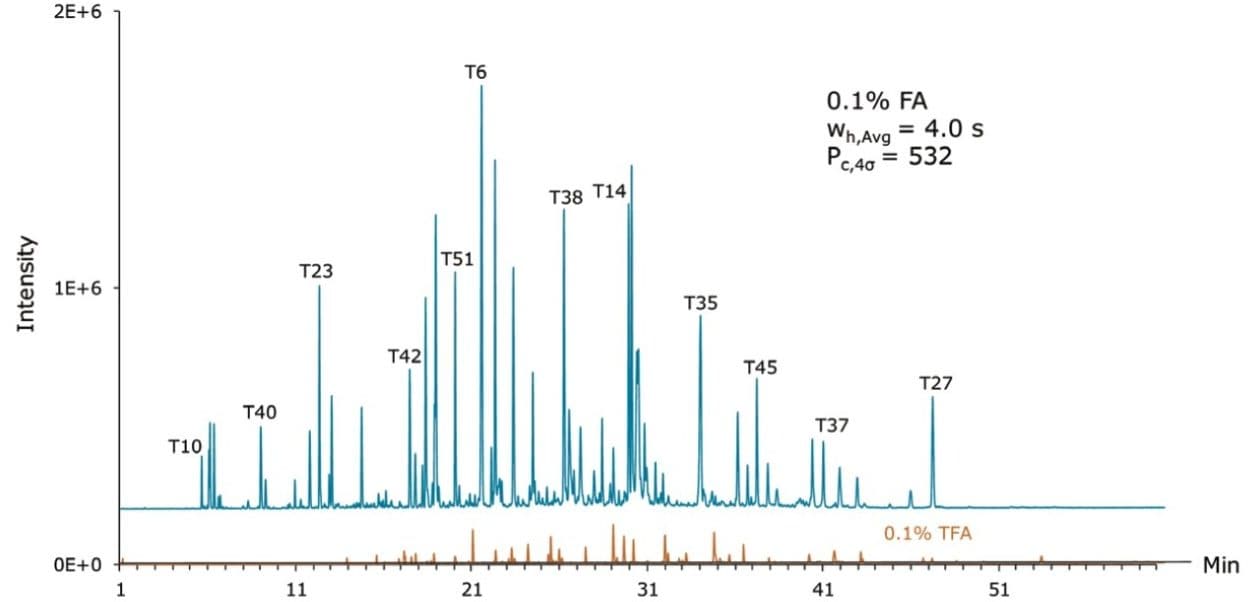
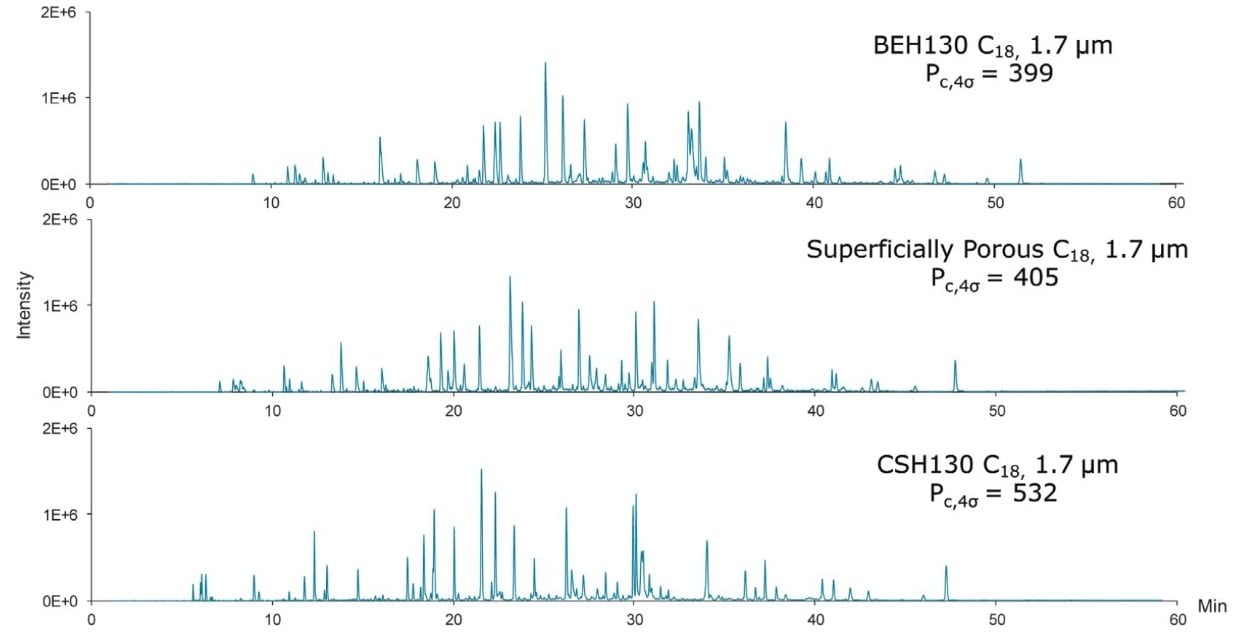
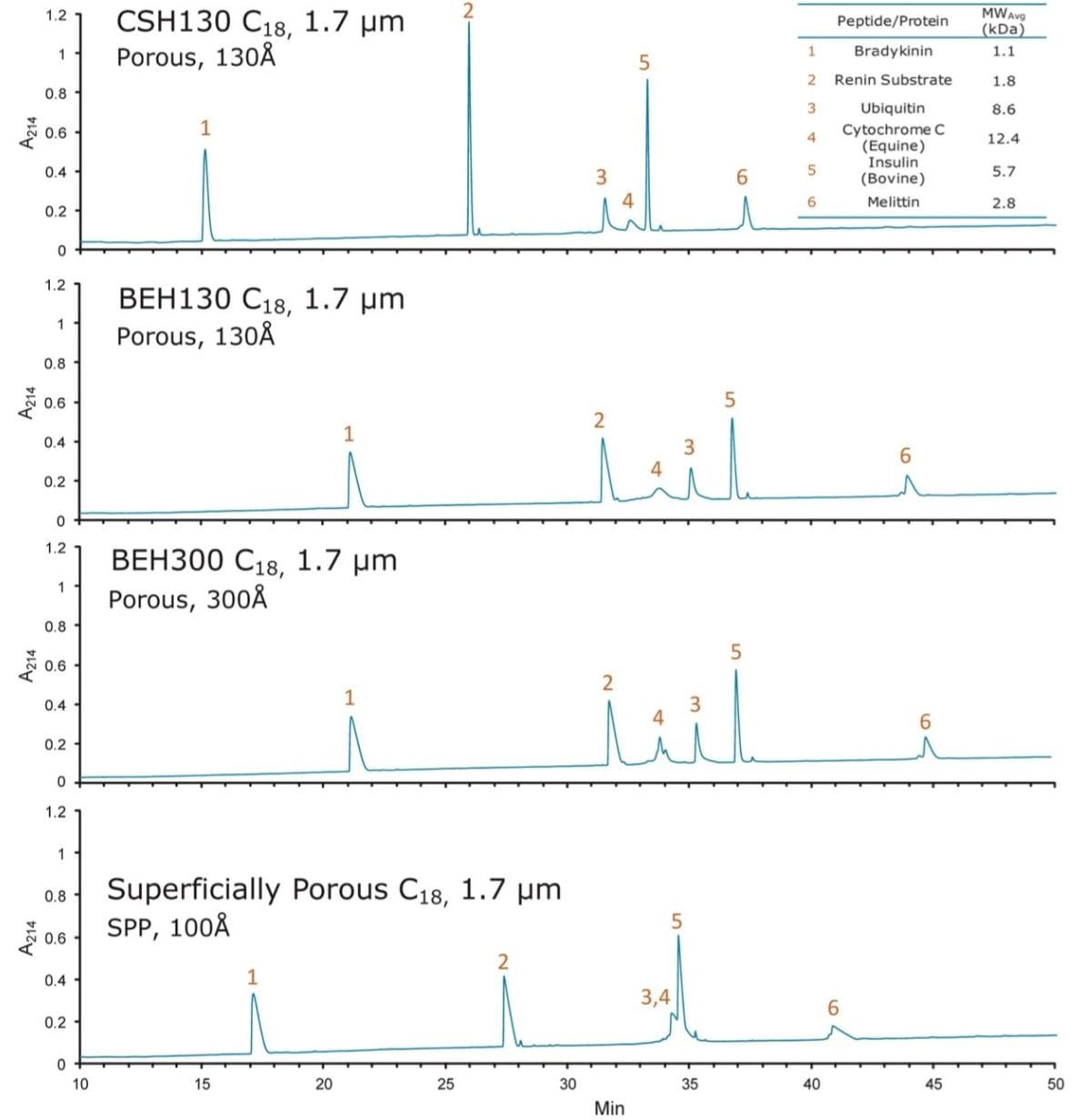
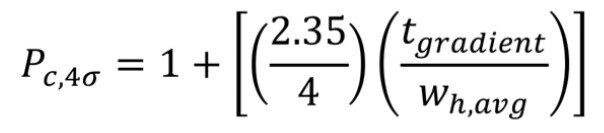Based on these measurements, the CSH130 C18, 1.7 µm column produced a peak capacity of 532, which is remarkably high for an LC-MS platform amenable to routine work. To provide perspective, the enolase digest was likewise analyzed by LC-MS using two 1.7 µm C18 columns that do not have a low level positive charge applied to the particle surface, as shown in Figure 2. The fully porous BEH130 C18, 1.7 µm column produced a peak capacity of 399, and the superficially porous C18, 1.7 µm column produced a similar peak capacity of 405. The novel CSH130 C18 stationary phase, thus, yielded a significant performance advantage for this application with 30% greater peak capacity.
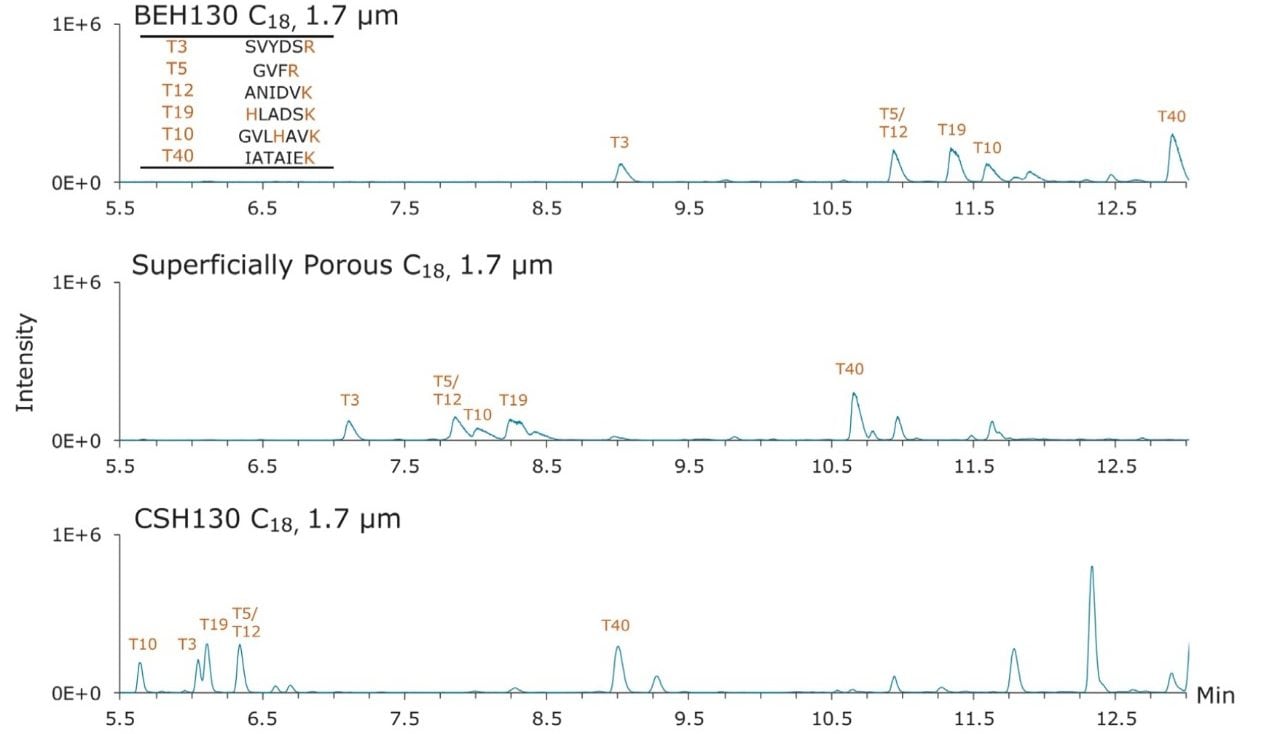
The retentivity and selectivity of peptides also varied between the three columns shown in Figure 2. An early time segment of the enolase peptide maps capturing this observation is shown in Figure 3. Peaks corresponding to six different peptides are labeled. The most immediate observations resulting from this comparison include: 1) CSH130 C18 provides better peak shape; and 2) CSH130 C18 is slightly less retentive than the other stationary phases. Elution of the labeled peptides from the CSH130 C18 column occurred approximately 5 min earlier compared to the BEH130 C18 column and approximately 2 min earlier compared to the superficially porous C18 column. In terms of elution strength, these are differences estimated at 4% and 2% acetonitrile, respectively. A more detailed analysis of these chromatograms shows the unique selectivity of the CSH130 C18 column. Elution order of the peptides changes quite dramatically when changing from the BEH130 C18 to CSH130 C18 column. Peptides with the largest selectivity differences in this chromatographic window appear to be peptides T10 and T19. Most tryptic peptides, such as T3, T5, T12, and T40, contain only two basic moieties, one N-terminus and one C-terminal lysine or arginine residue. Peptides T10 and T19, in contrast, also contain basic histidine residues, causing them to have an additional positive charge. This is most likely the reason for their relatively larger shift in retention time. The retention of peptides on CSH130 C18, therefore, seems to be influenced by their charge (or possibly their charge density), which in turn has an effect on selectivity. This suggests it is advantageous to screen both BEH130 C18 and CSH130 C18 columns when developing challenging peptide maps, particularly when aiming to separate critical pairs of peptides.