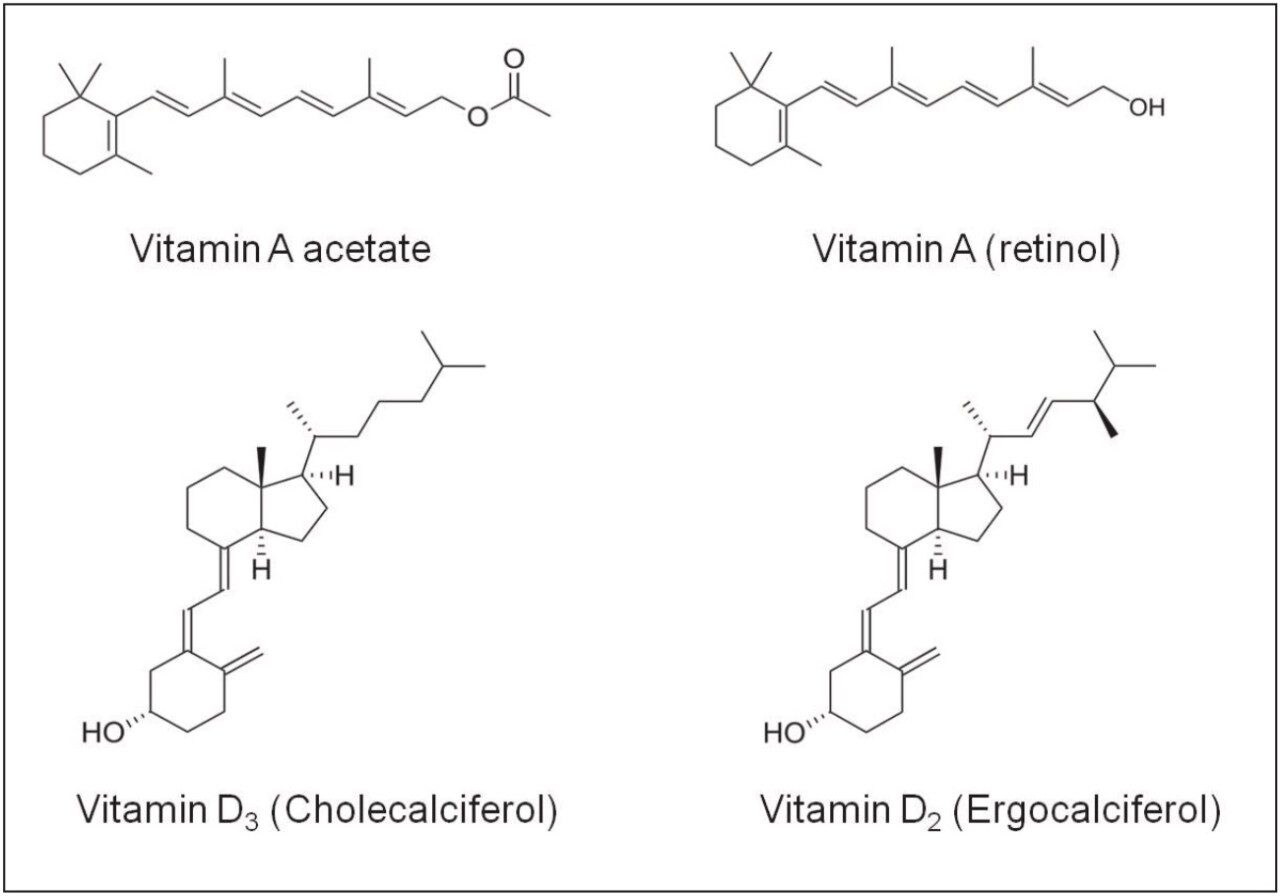
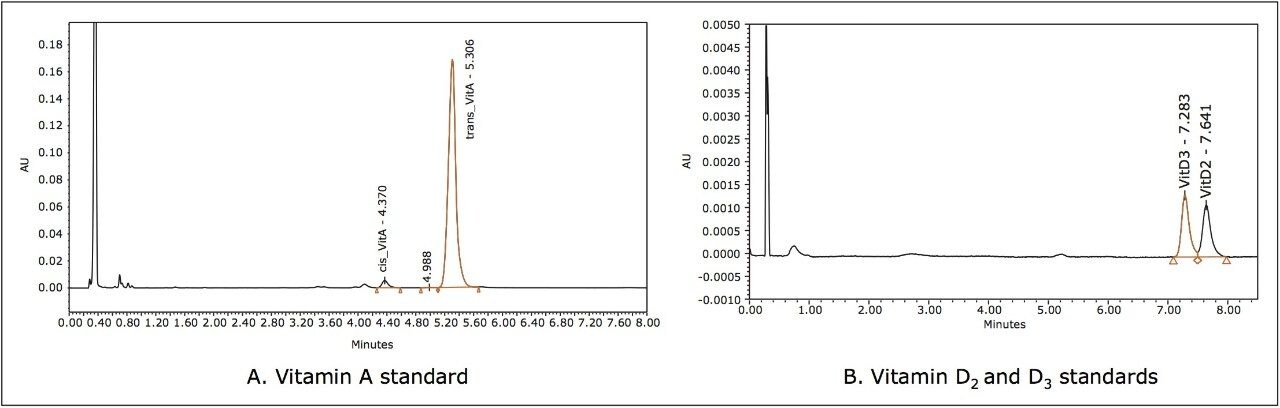
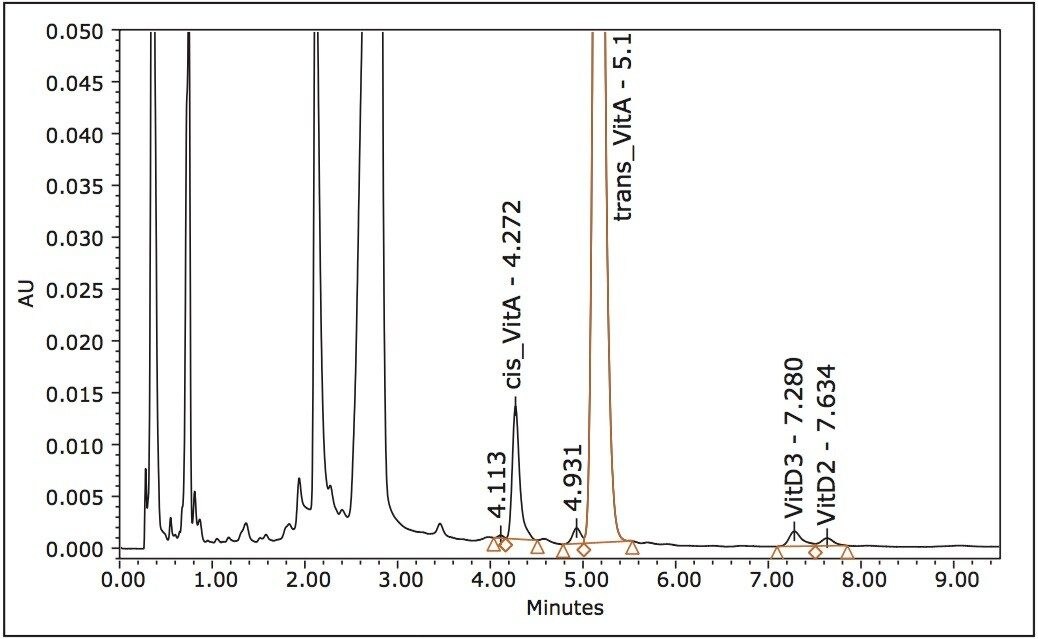
Simultaneous analysis of fat-soluble vitamins in foods is challenging due to their different properties and concentrations. A typical method involves extraction and saponification of fat, followed by high performance liquid chromatography (HPLC) with UV/Vis detection. After saponification the extracts can be analyzed for vitamin A directly, but they have to be diluted due to high abundance of the vitamin A in foods and its high molar extinction. Unfortunately, the dilution makes it impossible to detect vitamin D3 in the same solution. To measure vitamin D3, the extracts have to be cleaned on a semi-preparative chromatograph and concentrated. For these reasons vitamins A and D3 had to be analyzed separately. The HPLC of these compounds suffers from a long runtime, slow equilibration, and poor reproducibility.
As the extraction and saponification of separate vitamin A and D3 methods are identical, we investigated whether it would be possible to apply Waters UltraPerformance Convergence Chromatography (UPC2) to analyze extract for vitamin A and D3 in a single chromatographic run.
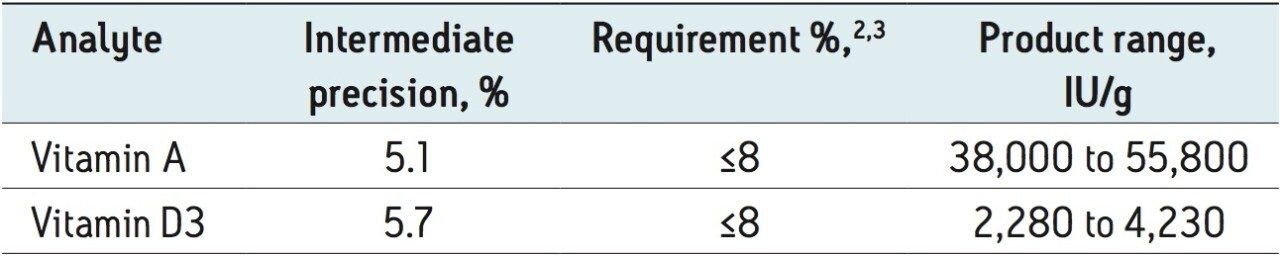
UPC2 is a separation technique that uses compressed carbon dioxide as the primary mobile phase. It takes advantage of sub-2 μm particle chromatography columns, the low viscosity of CO2, and an advanced chromatography system. This differs from traditional HPLC and improves the sensitivity of this assay. UPC2 also generates much less solvent waste compared to conventional liquid chromatography.1 In this application note, we report a method for analysis of vitamin A and D3 in vitamin premixes and concentrates in one analytical run without purification or dilution. The metrological properties of the UPC2 and the advantages of the method compared to the HPLC are also discussed.