Advanced Monoclonal Antibody Peptide Mapping Using Multi-Reflecting TOF MS Technology
Ce document est une note d’application et ne contient pas de section détaillée concernant l’expérimentation.
Abstract
Peptide mapping is a primary approach for the characterization and monitoring of biotherapeutics, particularly in the development of biopharmaceuticals such as monoclonal antibodies (mAbs). It enables detailed peptide level analysis to confirm protein identity and sequence and can localize any predicted modifications or product related impurities that could impact function. Furthermore, peptide mapping of a well characterized biotherapeutic can be a valuable tool for process monitoring and late development product commercialization.
In this application brief, we demonstrate the use of the ACQUITY™ Premier UPLC System coupled to the Xevo™ MRT Multi-reflectron Time-of-Flight (MRT) Mass Spectrometer, which incorporates novel multi reflecting time of flight technology, for the analysis of the Waters™ mAb tryptic digestion standard. Characterization was performed with the peptide mapping workflow in the waters_connect UNIFI™ App. The results highlight the Xevo MRT MS platform's high sensitivity, high mass resolution and consistent sub-ppm mass accuracy, along with integrated waters_connect data acquisition and processing capabilities. This enables highly confident characterization capabilities enabling faster and better product and process development decisions.
Benefits
- High levels of sensitivity, allowing in depth mAb characterisation and detection of low-level PTMs and peptide attributes
- Dynamic range of 5 orders of magnitude enabling analysis of complex samples with product variants and degradants easily identified at lower levels
- Consistent mass error below 1 ppm, ensuring confident product and impurity identifications
Introduction
Peptide-digest level analysis of monoclonal antibodies (mAb) is a primary analytical method used throughout the lifecycle of therapeutic mAbs to ensure product quality and consistency. As biotherapeutic pipelines expand, it becomes increasingly important to deploy high confidence automated methods that can accurately and consistently characterize low-level mAb modifications for efficient product and process development decision making. This requires robust, easy-to-use analytical platforms that can be readily deployed in laboratories across an organization.1
The Xevo MRT MS System exemplifies such a platform This system uses a novel quadrupole-multi reflecting time-of-flight design to extend ion flight paths, resulting in mass resolution of up to 100K at a 50 Hz scan rate, and a dual gain ADC detector that enables greater sensitivity, dynamic range, and sub ppm mass accuracy for confident peptide mapping. The compliance ready waters_connect software with the UNIFI App provides automated data acquisition, processing and reporting, while integrated and automated workflows enhance the accessibility of LC-MS analyses as molecules move beyond the MS core labs focussing on initial and extended product characterization.
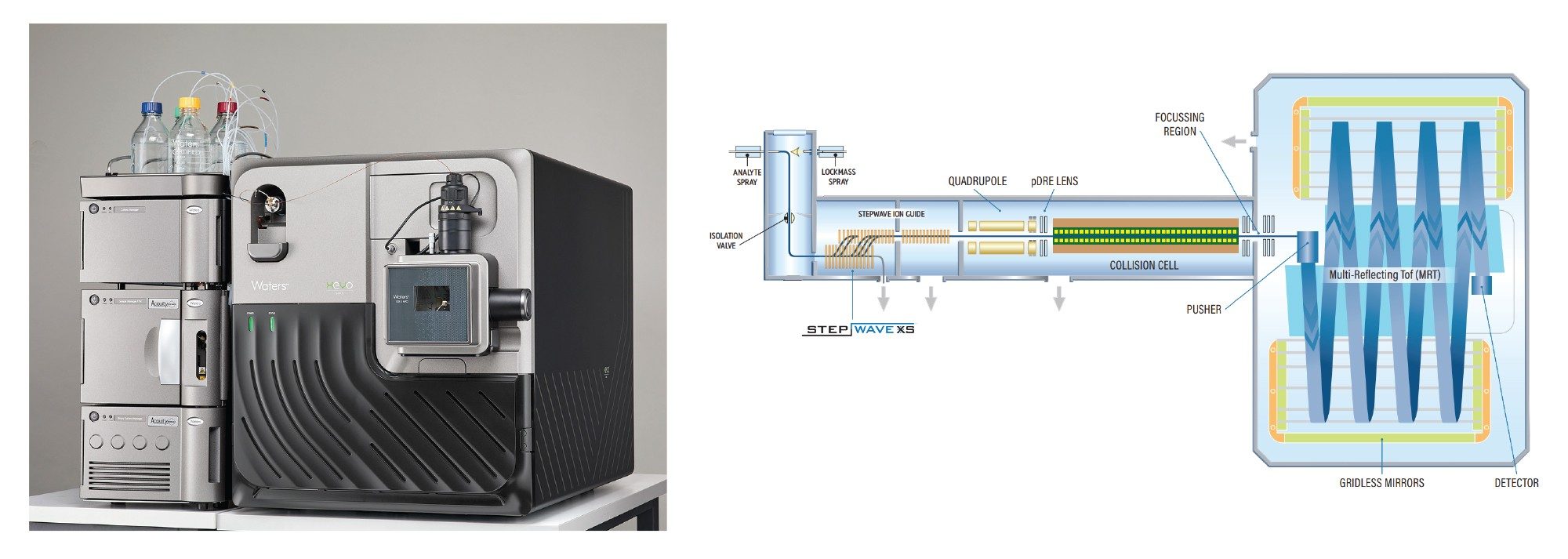 Figure 1. Xevo MRT MS System.
Figure 1. Xevo MRT MS System.
Experimental
Sample
The Waters mAb Tryptic Digestion Standard (p/n: 186009126) was used for benchmarking LC-MS performance and ensuring system suitability when performing peptide-level protein analysis. This lyophilized standard contains a reduced and alkylated tryptic digest of NIST Reference Material 8671 (NIST mAb), a humanized IgG1κ expressed from a murine cell line.
To prepare the mAb sample, 200 µL of ultrapure Milli-Q H2O was added to the 40 µg digest vial, resulting in a concentration of 200 ng/µL. This sample was then further diluted to provide a range of concentrations, allowing column loadings between 2000 ng and 0.002 ng to be evaluated.
LC Conditions
|
LC system: |
ACQUITY Premier UPLC System (Binary) |
|
Detection: |
UV 280 nm |
|
Vials: |
TruView™ LCMS Certified 12 x 32 mm Screw Neck Vial, Total Recovery, with Cap and Preslit PTFE/Silicone Septum (p/n: 186005663CV) |
|
Column: |
ACQUITY Premier BEH™ C18, 1.7 µm 2.1 mm x 100 mm (p/n: 186009453) |
|
Column temperature: |
40.0 °C |
|
Sample temperature: |
6.0 °C |
|
Injection volume: |
0.2 to 10 µL |
|
Flow rate: |
0.250 ml/min |
|
Mobile phase A: |
H2O 0.1% formic acid |
|
Mobile phase B: |
ACN 0.1% formic acid |
Gradient Table
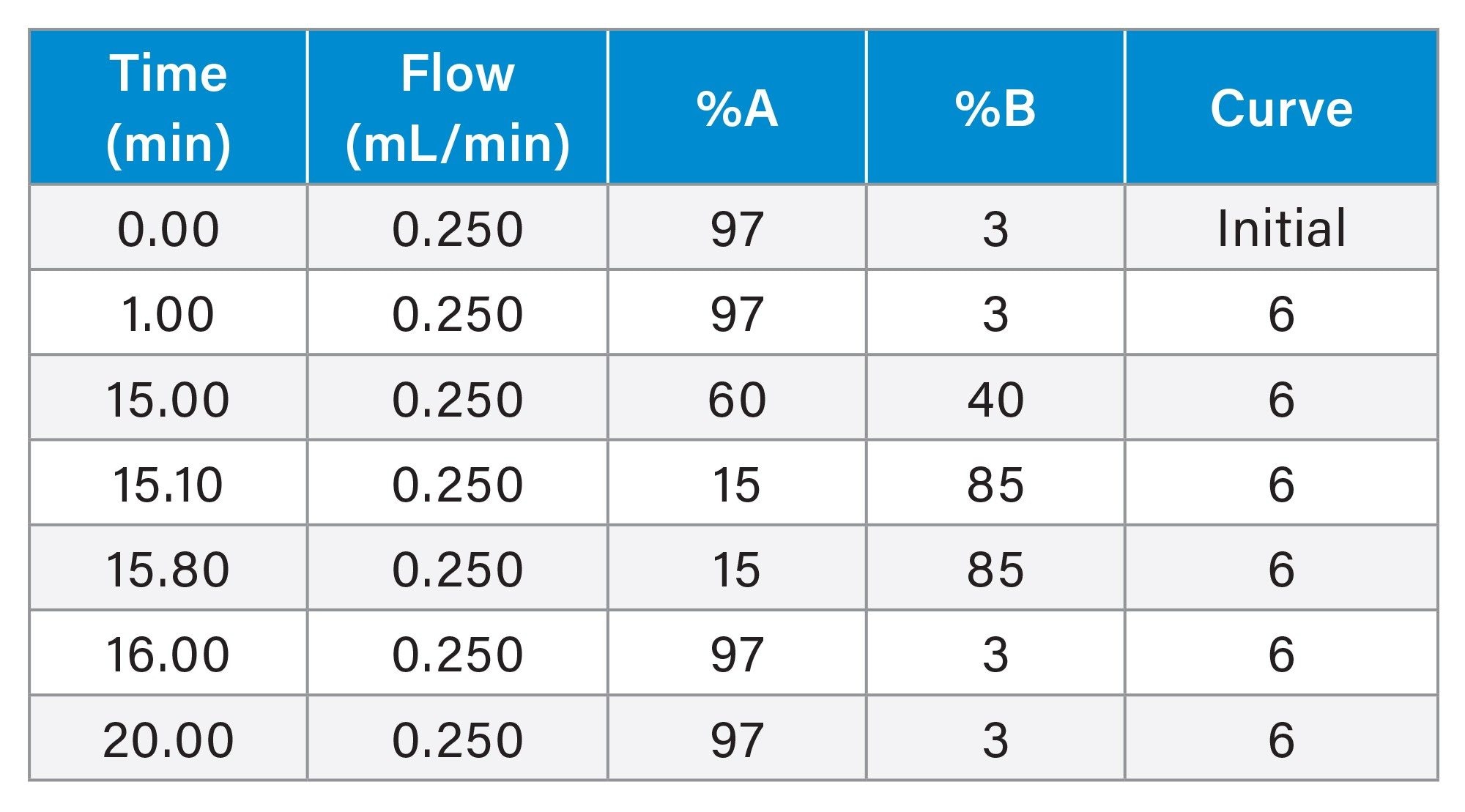
MS Conditions
|
MS system: |
Xevo MRT MS |
|
Mode: |
MSE Full scan with fragmentation |
|
Mass range: |
50–2000 m/z |
|
Polarity: |
+ve |
|
Scan rate: |
2 Hz |
|
Cone voltage: |
25 V |
|
Fragmentation collision energy ramp: |
25–50 V |
|
Source temperature: |
120 °C |
|
Desolvation temperature: |
400 °C |
|
Desolvation gas: |
800 L/hr |
|
Capillary voltage: |
3.00 kV |
Results and Discussion
Peptide mapping data on a NIST mAb digested standard was acquired on the UPLC-Xevo MRT MS System to assess overall workflow performance and report specific instrument capabilities for mass accuracy, mass resolution and sensitivity. Data were acquired in MSE mode, with data independent fragmentation, to assign peptides and localize sites of assigned modifications.
High sequence coverage (96% LC, 98% HC) was achieved for both the light and heavy chains with less than 1 ppm mass error tolerances from automated data processing within the waters_connect UNIFI App peptide mapping workflow. Figures 2, 3, and 4 contain the output results from the peptide mapping experiments.
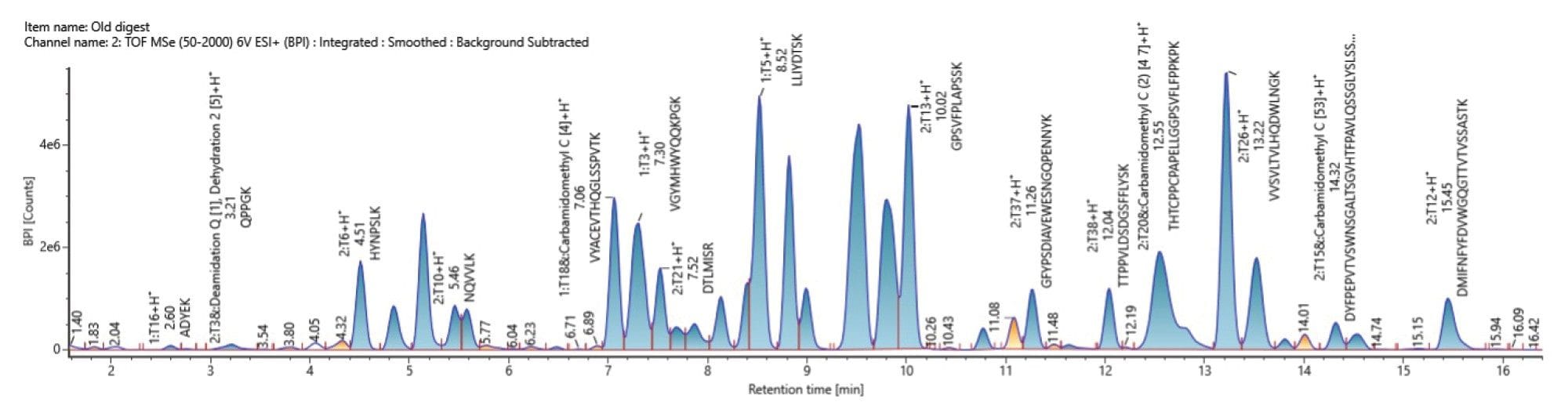 Figure 2. A processed Base Peak Intensity (BPI) chromatogram of NIST mAb digest standard peptides.
Figure 2. A processed Base Peak Intensity (BPI) chromatogram of NIST mAb digest standard peptides.
 Figure 3. The Sequence coverage of mAb Tryptic Digestion Standard based on aa with sub-ppm mass tolerance (96% sequence coverage for light chain, 98% for heavy chain).
Figure 3. The Sequence coverage of mAb Tryptic Digestion Standard based on aa with sub-ppm mass tolerance (96% sequence coverage for light chain, 98% for heavy chain).
Shown in Figure 4, The list of assigned peptides was produced by comparison against an in-silico digest of the NIST mAb, also accounting for modifications defined in the method. This list can be filtered and ranked by intensity, mass tolerance, and the number of matched fragments. The high-ranking assigned peptides all have less than 1 ppm mass error and are further confirmed by a high number of matched b/y fragments obtained from the data-independent MSE fragmentation.2,3,4
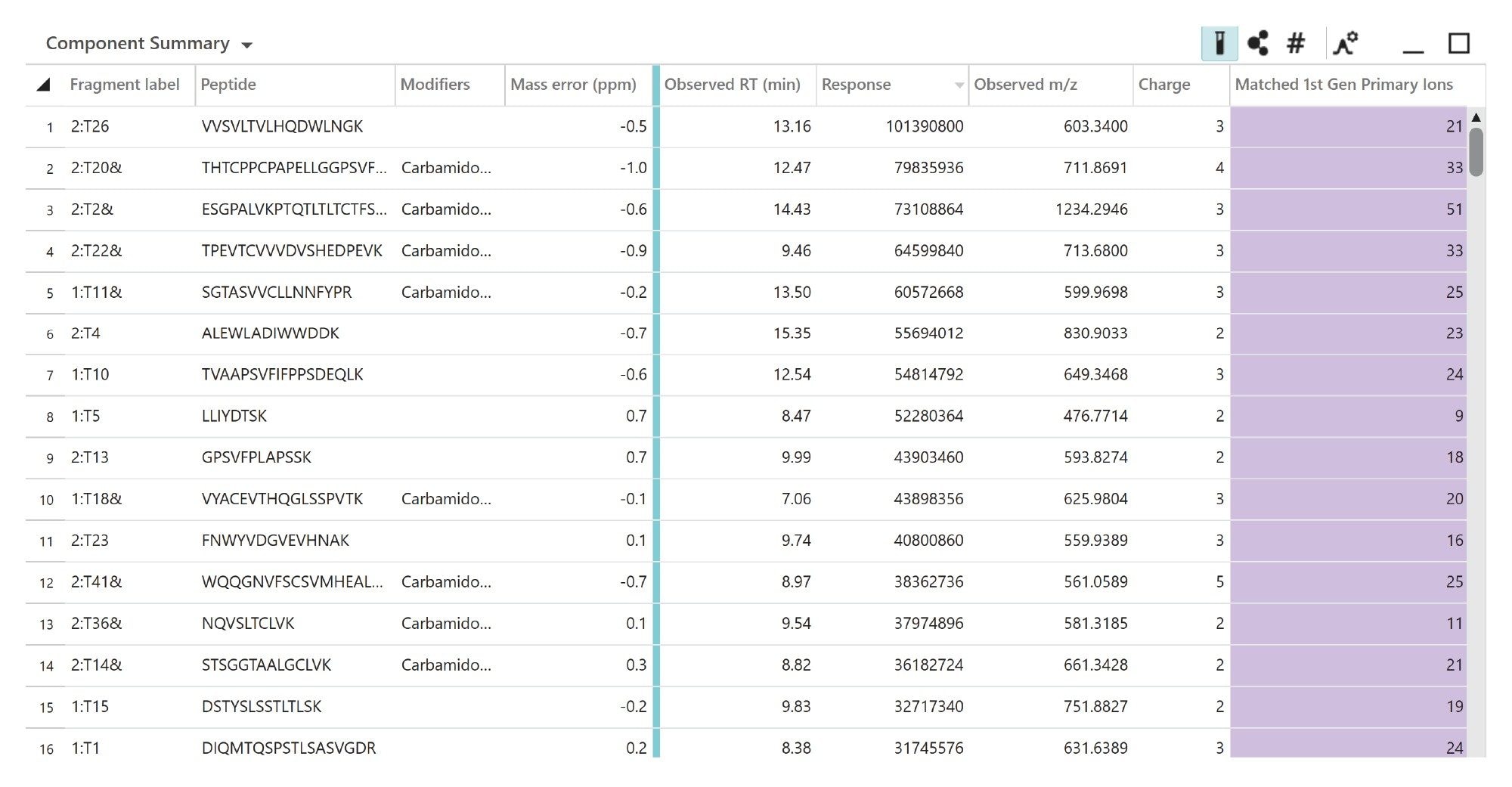 Figure 4. UNIFI peptide mapping identification results <1 ppm.
Figure 4. UNIFI peptide mapping identification results <1 ppm.
Utilising MSE for the analysis provides a comprehensive data set for peptide map based characterization of proteins. Unlike traditional data-dependent acquisition methods, MSE employs data-independent acquisition, capturing MS2 fragment ions for all MS1 peptide ions in a single run. This approach provides both high sequence coverage supported by accurate mass fragmentation data and accurate quantification from MS1 peak quantification, all within a single injection. The ability to routinely and reproducibly generate precursor and fragment data makes the MSE approach ideal for assignment of peptide ions and relative quantitation of product related variants and impurities for the protein drug product or substance. The unbiased nature of MSE ensures that even lower-abundance peptides can be confirmed with characteristic fragments, enhancing the depth and reproducibility of mAb characterisation.
Figure 5 highlights the DTLMISR peptide, both its native and oxidized variants are observed with sub ppm mass accuracy, The oxidized variant of the DTLMISR peptide elutes earlier (RT 6.48 minutes) compared to the unmodified peptide (RT 7.52 minutes). The high quality MSE Fragmentation spectra identify a +15.9949 Da mass shift on the y4, y5 and precursor indicating the oxidation has taken place at the Methionine residue.
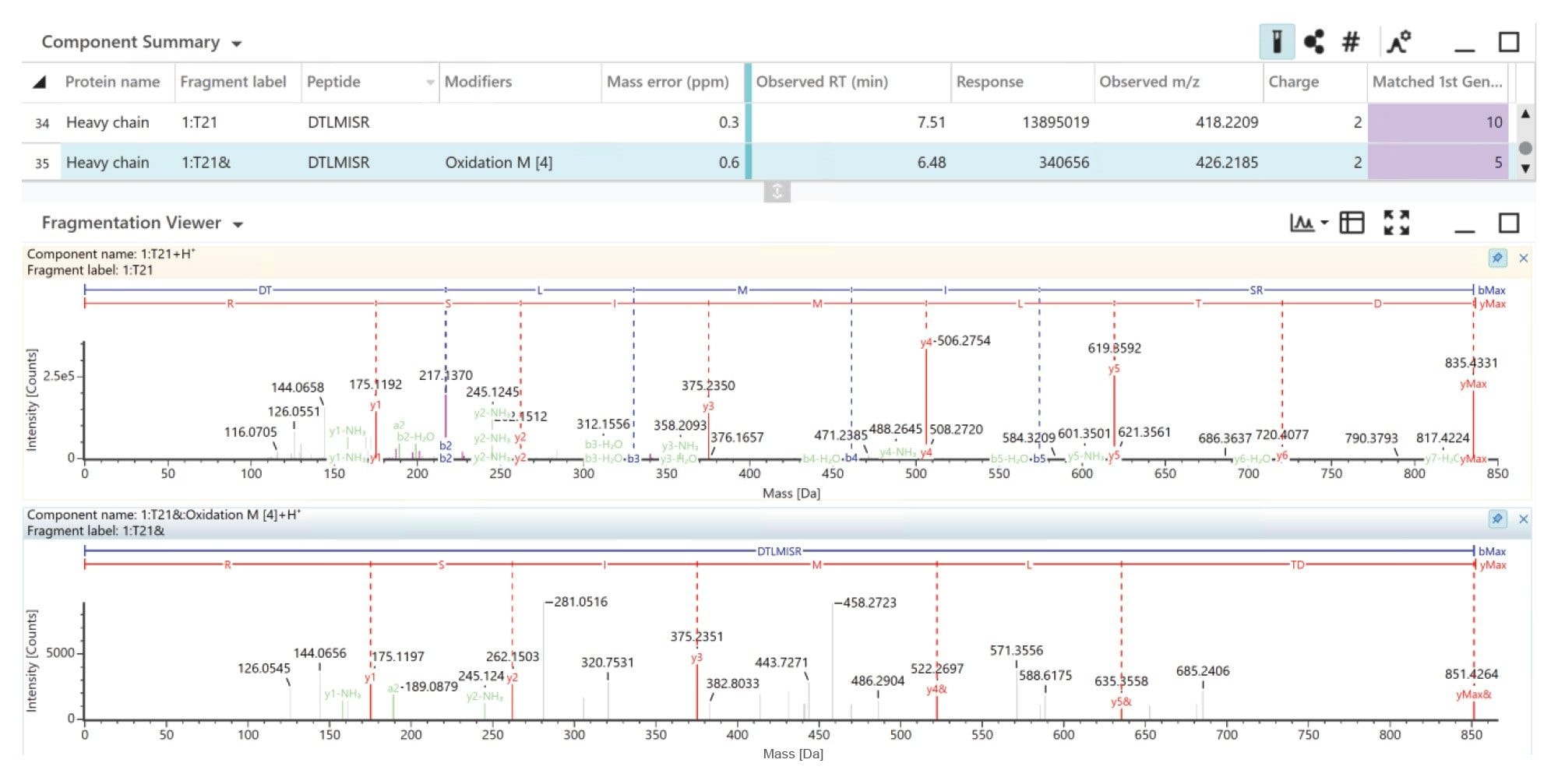 Figure 5. UNIFI identification and MSE spectra of oxidized (upper) and unmodified (bottom) DTLMISR peptide.
Figure 5. UNIFI identification and MSE spectra of oxidized (upper) and unmodified (bottom) DTLMISR peptide.
The T25 glycopeptide (G0F) response from ten replicate injections (Figure 6) shows overlaid chromatograms with near-perfect overlap, demonstrating the inherent reproducibility of the data-independent LC-MSE peptide mapping approach. The chromatograms show consistent UPLC chromatography and consistent LC-MS detection response across the ten injections. Figure 7 further expands on this showing the summed MS1 response of HC T25 glycopeptide G0F across replicate injections, demonstrating an intensity RSD of 1.5%.
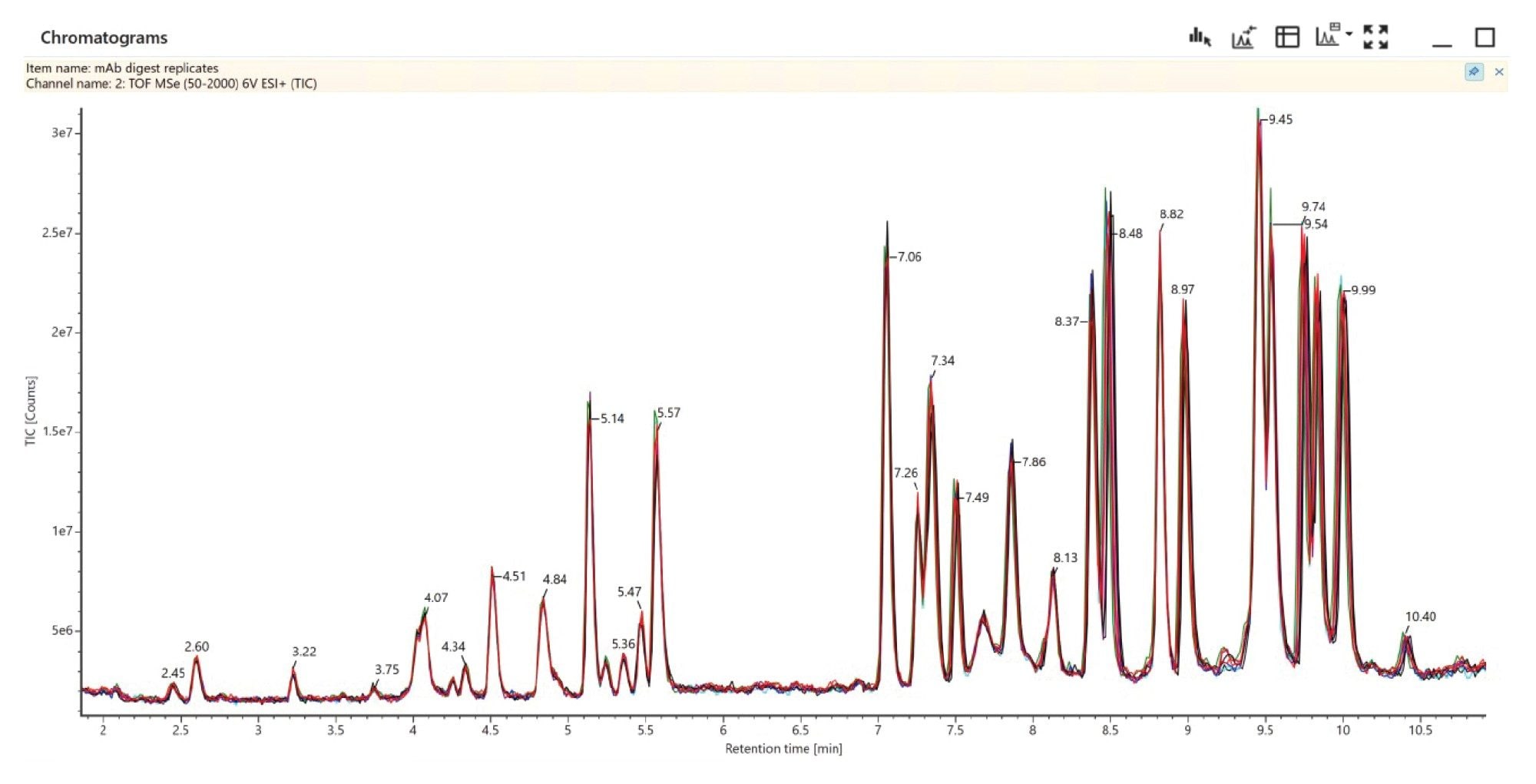 Figure 6. TOF TIC Data from ten replicate NIST mAb digest injections, overlaid, demonstrating reproducibility both in chromatography and MS response.
Figure 6. TOF TIC Data from ten replicate NIST mAb digest injections, overlaid, demonstrating reproducibility both in chromatography and MS response.
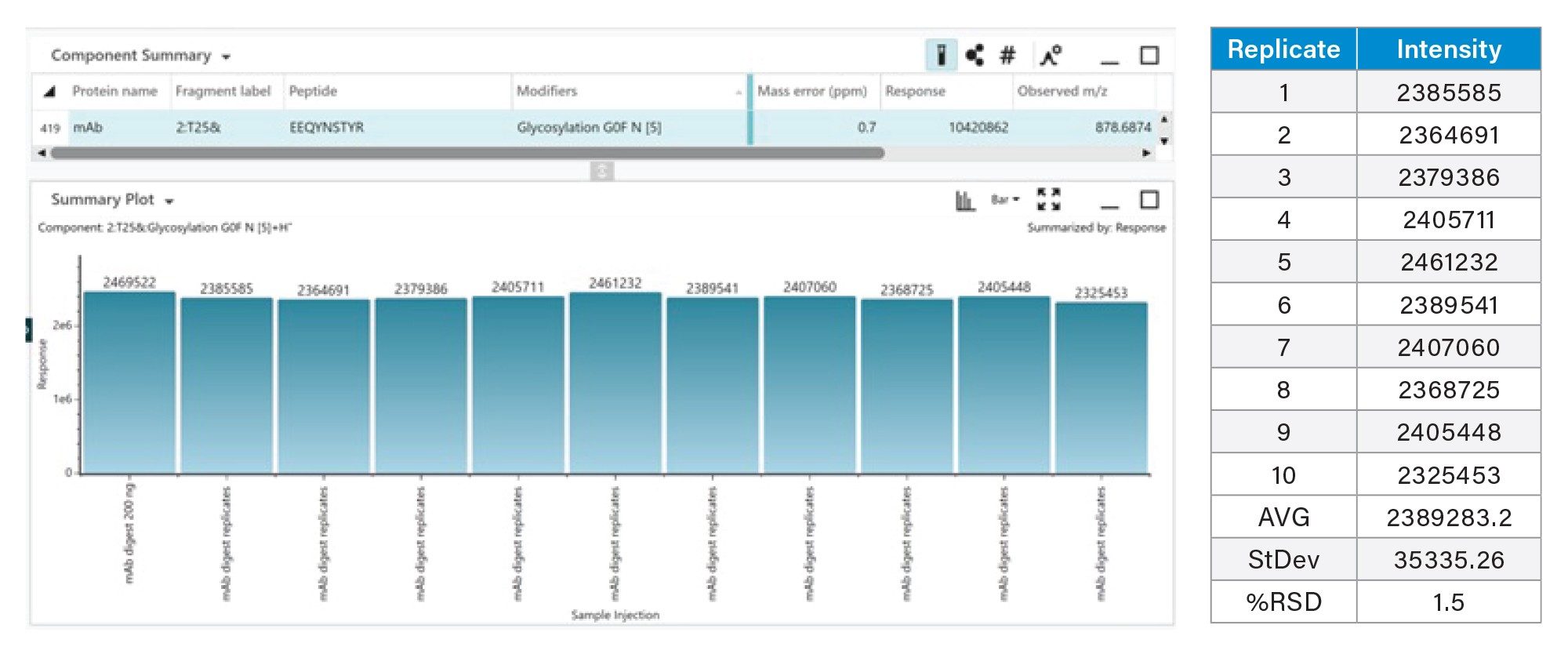 Figure 7. Summed MS1 response of HC T25 glycomodified G0F peptide across replicate injections, demonstrating an intensity RSD of 1.5%.
Figure 7. Summed MS1 response of HC T25 glycomodified G0F peptide across replicate injections, demonstrating an intensity RSD of 1.5%.
At least four orders of dynamic range are needed for comprehensive protein impurity analysis, as it allows for differing ionization potential of tryptic peptides, and the detection and quantification of modified peptides across a wide relative concentration. The dilution series in Figure 8 shows a selected peptides extracted ion chromatograms at digest column loadings of 2000 ng, 2 ng, and 0.02 ng (five orders) of the mAb digestion standard along with a demonstrated linear MS1 response between 1000 ng and 0.02 ng.
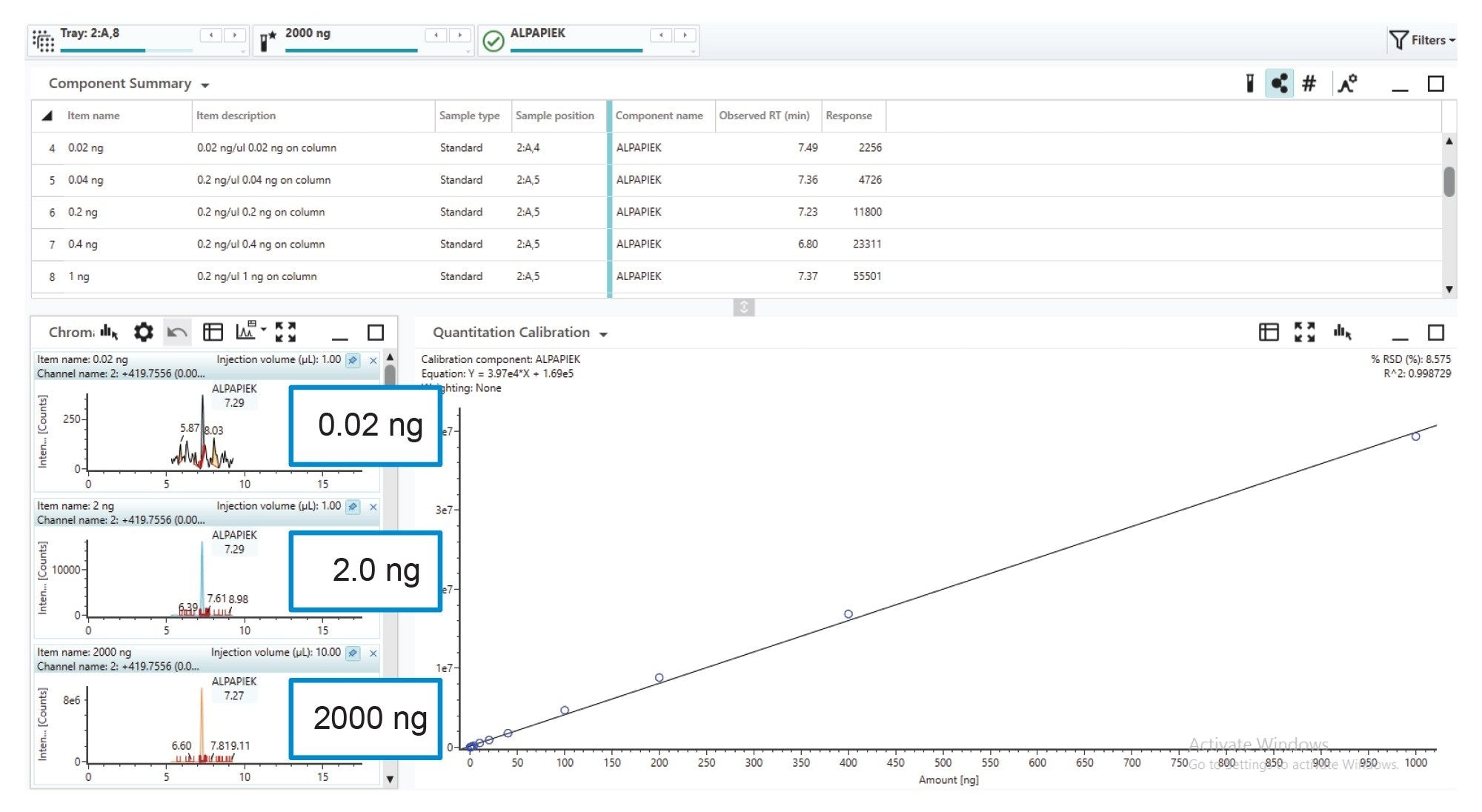 Figure 8. Quantitation results for a selected peptide (ALPEPIEK), demonstrate dynamic range of quantification spanning five orders of magnitude with R² (0.999) and RSD (<10%) fit values obtained for the linear calibration. High sensitivity and linear MS response enable confident tracking of lower-level CQA’s across samples.
Figure 8. Quantitation results for a selected peptide (ALPEPIEK), demonstrate dynamic range of quantification spanning five orders of magnitude with R² (0.999) and RSD (<10%) fit values obtained for the linear calibration. High sensitivity and linear MS response enable confident tracking of lower-level CQA’s across samples.
Localizing and profiling glycosylation sites is essential for understanding the biological activity, safety profile and therapeutic efficacy of monoclonal antibodies (mAbs). Here, we demonstrate how the peptide mapping workflow with the Xevo MRT MS System provides consistent glycopeptide MS response for the NIST mAb standard, as shown in Figure 9 for glycopeptides assigned with less than 1 ppm mass error. This consistency enables us to also observe the trend changes in undesirable Man5 levels in the mAb between two mAb batches for the highlighted T25 Man5 glycopeptide.
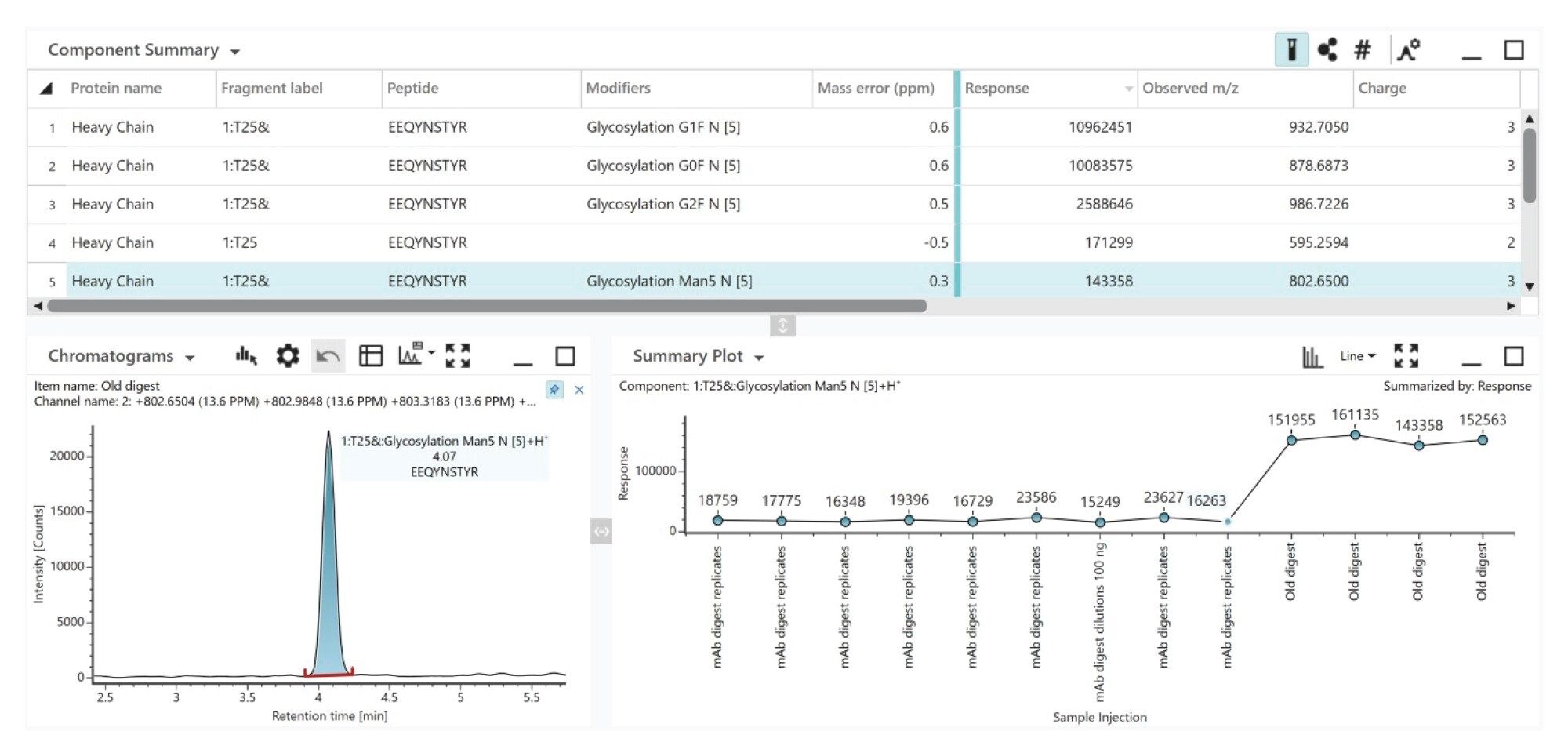 Figure 9. The peptide mapping workflow of the Xevo MRT MS System enables confident assessment of sample-to-sample changes in mAb glycosylation. The figure shows the data review panel in the UNIFI App following automated data acquisition and processing of replicate data from digests of two mAb batches. The component table contains sequence information, m/z and neutral mass, mass tolerance, %rel. abundance as well as fragmentation information for each PTM. The summary plot provides a concise view of MS1 intensity for the early eluting the Man5 glycopeptide peak observed across all samples.
Figure 9. The peptide mapping workflow of the Xevo MRT MS System enables confident assessment of sample-to-sample changes in mAb glycosylation. The figure shows the data review panel in the UNIFI App following automated data acquisition and processing of replicate data from digests of two mAb batches. The component table contains sequence information, m/z and neutral mass, mass tolerance, %rel. abundance as well as fragmentation information for each PTM. The summary plot provides a concise view of MS1 intensity for the early eluting the Man5 glycopeptide peak observed across all samples.
Conclusion
The Xevo MRT Mass Spectrometer offers numerous benefits for peptide-based biotherapeutic characterization, providing high confidence sequence confirmation and assignments of product variants and impurities. The high performance multi-reflecting time-of-flight (MRT) technology delivers sub-ppm mass accuracy for confident peptide assignments from both precursor and fragmentation data, and an effective mass resolution approaching 100,000 FWHM that is generated for peptide ions independent of scan speed and m/z, ensuring precise and accurate analyte identification across the map.
This exceptional MS detection performance was complemented with a streamlined automated compliant-ready workflow, within the UNIFI App on the waters_connect informatics platform that supported integrated data acquisition, processing, review, and reporting for peptide mapping data from individual samples, and across larger sets of analysed samples.
Overall, the Xevo MRT Mass Spectrometer was demonstrated to provide a robust and efficient platform for achieving reliable experimental outcomes that can enable biopharmaceutical analysts the data to make confident decisions on biotherapeutic development and commercialization.
For more information, please visit Xevo MRT Mass Spectrometer
References
- European Medicines Agency (EMA). (2016). Guideline on development, production, characterisation and specification for monoclonal antibodies and related products. EMA/CHMP/BWP/532517/2008. EMA website.
- Nilini Ranbaduge, Henry Shion, Ying Qing Yu. Routine Peptide Mapping Analysis using the BioAccord System. Waters Application Note 720006466. 2019.
- Kellen DeLaney, Samantha Ippoliti, Lisa Reid, Owen Cornwell, Ying Qing Yu, Emma Harry, Mark Towers. Applying Peptide Mapping and Multi-Attribute Method (MAM) Workflow for Biosimilar mAb Drug Products Comparison on the Xevo™ G3 QTof Platform Waters Application Note 720007632. 2022.
- Vera B. Ivleva, Ying Qing Yu, Scott J. Berger, Weibin Chen. Generating Automated and Efficient LC-MS Peptide Mapping Results with the Biopharmaceutical Platform Solution with UNIFI Waters Application Note 720004620. 2013.
Featured Products
720008737, March 2025