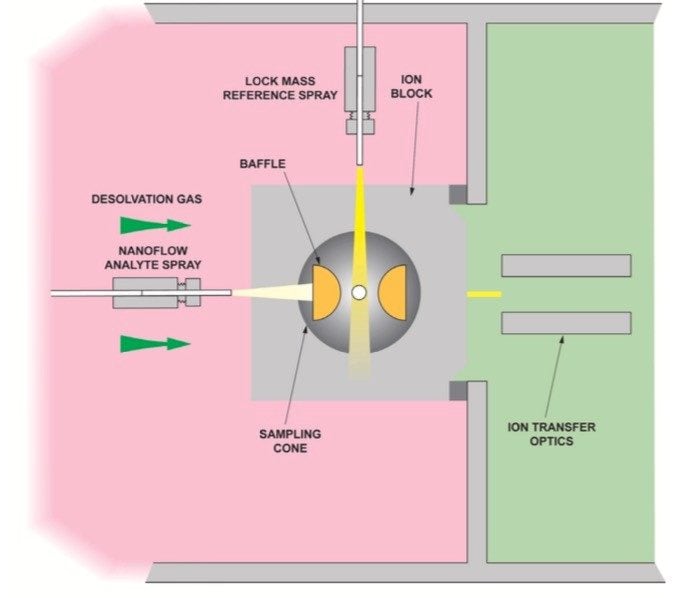
Mass Measurement Accuracy and de novo sequencing
A challenge remains in an LC-MS/MS experiment in that often many of the MS/MS spectra acquired do not provide matches when searched against known protein sequence databanks. The nature of database searching is such that only peptides that match exactly those within the databank will be identified. Consequently, many good quality spectra of novel peptides or of those containing a single amino acid substitution, or modification remain unmatched. Similarly, samples from organisms poorly represented in sequence databases can often produce a large number of unidentified spectra. Currently a common solution to this would be to extract these spectra manually from the data set, derive some degree of peptide sequence and perform further database searching. This, however, can be time consuming when large numbers of spectra are involved and is reliant upon the skill of the operator.
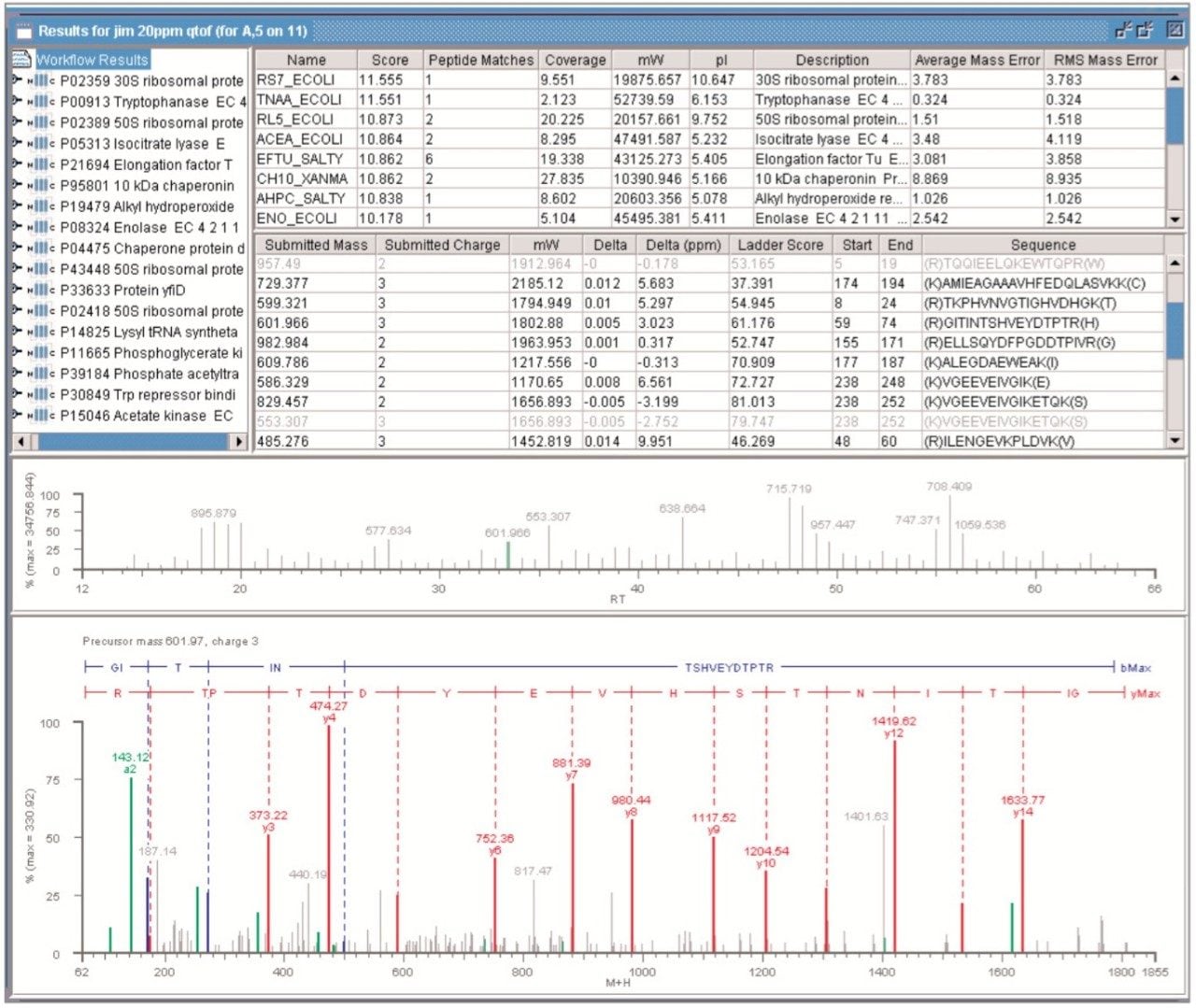
With the introduction of an automated computer-sequencing algorithm, MS/MS spectra can be identified by direct derivation of the novel peptide sequence through a double application of a Bayesian probabilistic analysis. A fragmentation model is applied to compare trial sequences against singly charged, de-isotoped MS/MS spectra. An overall confidence value for the most probable sequence, along with a confidence in the assignment of each individual residue, is calculated.
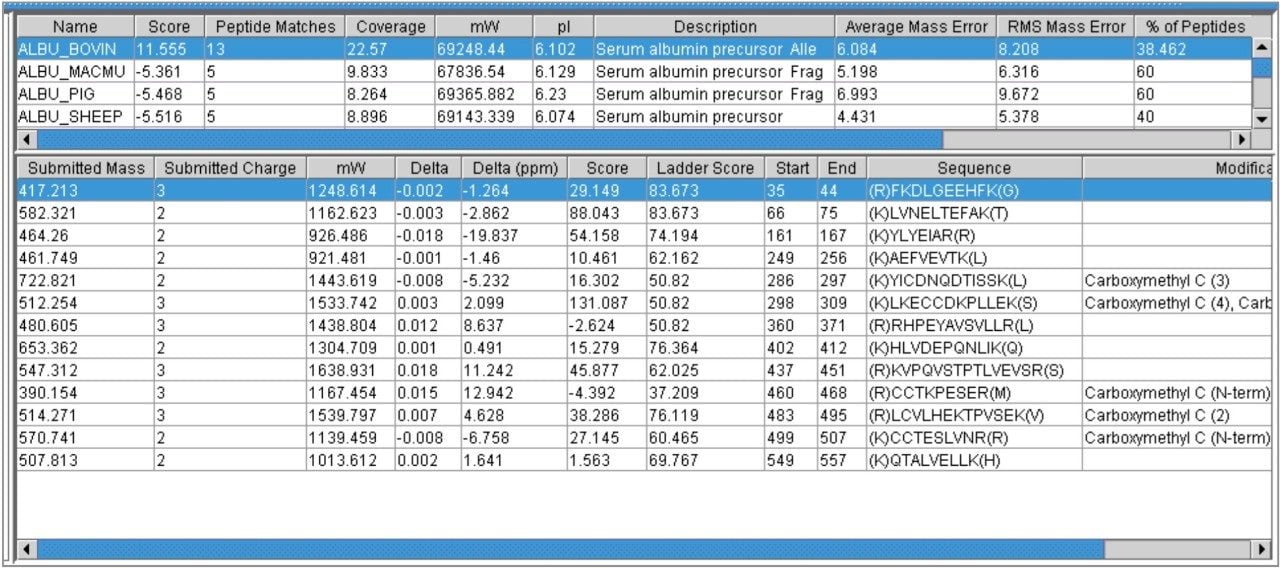
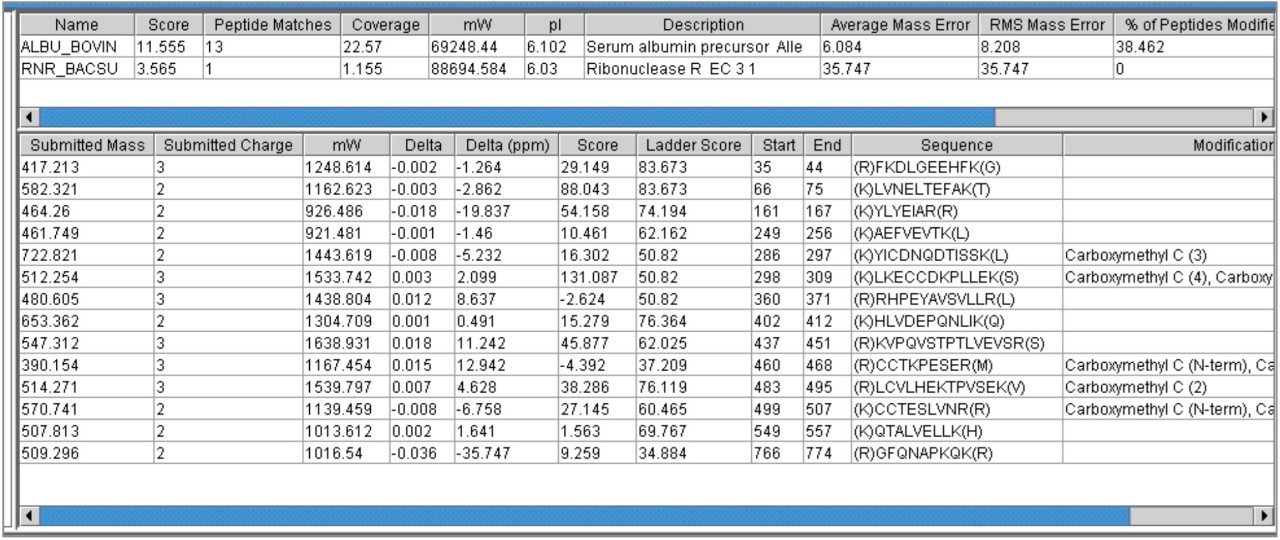
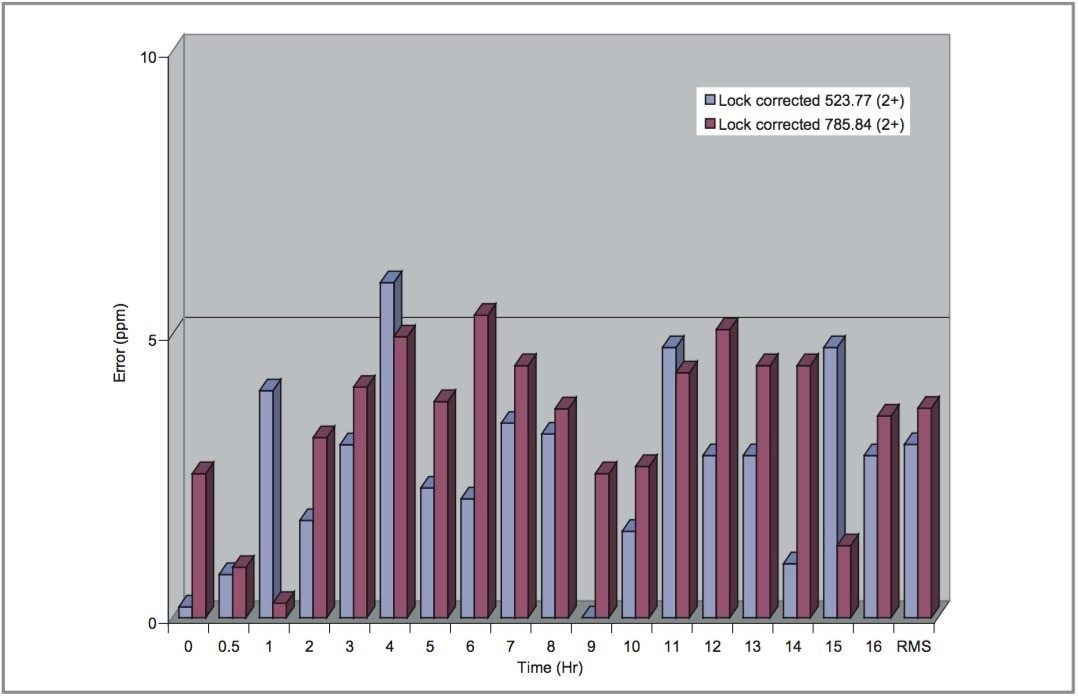
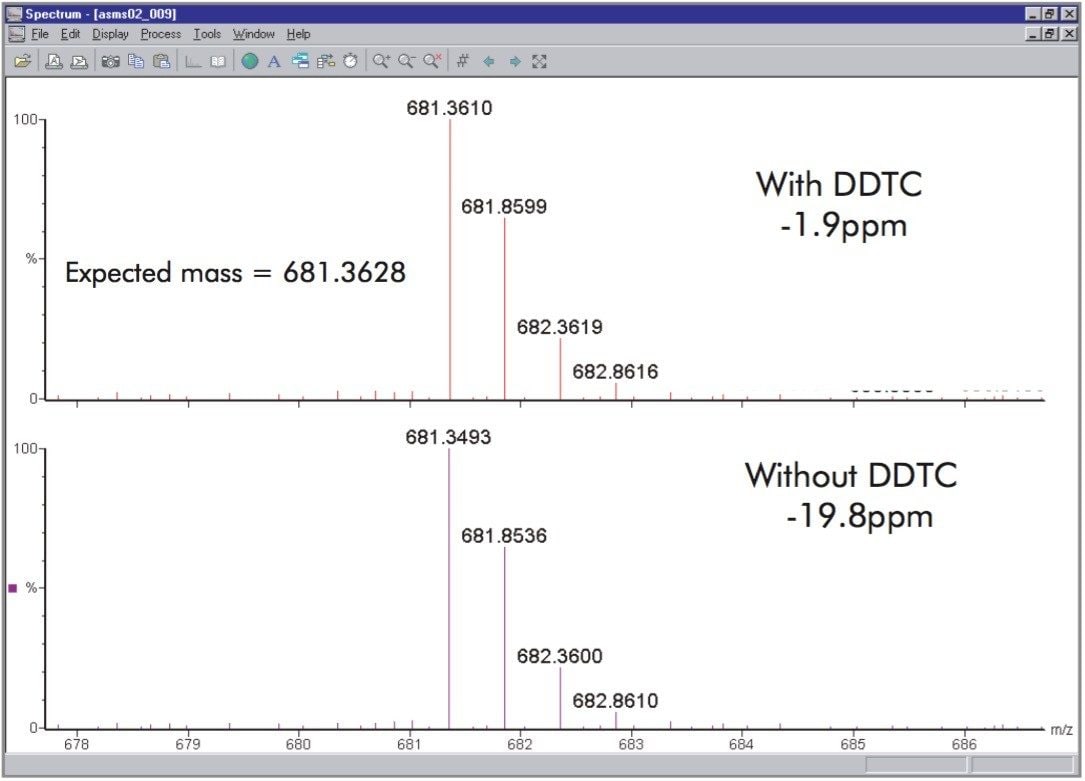
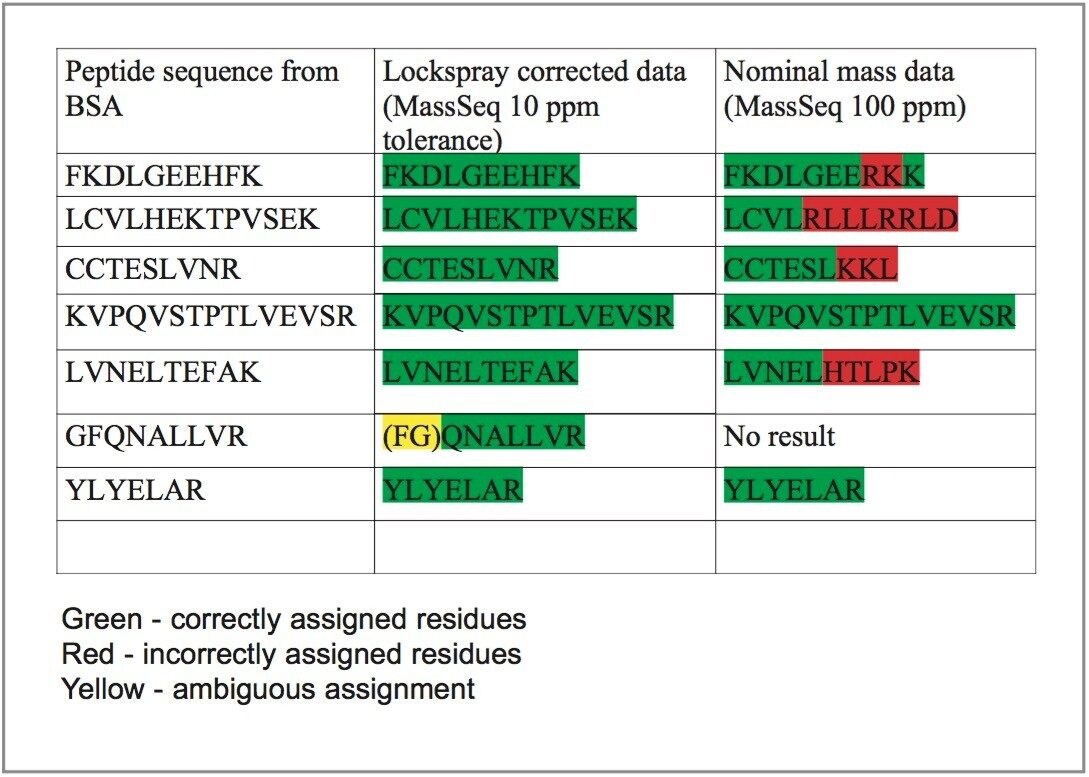
The MassSeq algorithm (ProteinLynx Global SERVER v2.0) utilizes the exact mass measurement of the MS/MS fragment ions to confidently define the most probable amino acid sequence. This is illustrated in Figure 8 where MS/MS spectra were acquired at different mass measurement accuracy using an ESI Q-Tof. It can be seen, in the resulting sequence determinations, that the data measured to less than 10 ppm provides significantly better results than that acquired at 100 ppm.