甜菊是一种原产于南美洲的菊科常青灌木。由于其叶片中富含具有食用价值的天然甜味剂,甜菊具有重要的经济价值。它又被称为“巴拉圭甜草”。目前,北美洲、南美洲、亚洲和一些欧洲国家已将甜菊植株或提取物用作甜味剂。食品添加剂联合专家委员会(JECFA)建立了一系列关于甜菊糖苷的规定,要求七种化学定义的甜菊糖苷必须达到95%以上的纯度1。
对食品中的甜菊糖苷进行全面的残留分析是一项颇为棘手的任务,因为此类分析需要利用通用提取步骤检测复杂食品类商品中的低浓度甜菊糖苷异构体,其本身具有一定的局限性。该分析存在两方面的挑战,一是样品中的甜菊糖苷含量可能很低,因为它们可能会与其它甜味剂或糖类共同使用。此外,如果样品中的一种或两种糖苷(如莱苞迪甙A)的浓度极高,则进行低浓度检测将非常困难。化合物的异构体可能具有不同的化学性质 — 它们可能风味各异,吸收、分布、代谢特性、去除方法和毒性也不尽相同。因此,我们有必要了解能够决定食品风味的物质组成。我们可采用一种具有高选择性和高灵敏度的初步筛选方法进行定性分析,除此之外还需要进行大量的检测工作以测定甜味剂的纯度,因为纯度也会影响风味2-4。
高分辨率质谱(HRMS)全扫描具有高度特异性,理论上其可检测的化合物数量没有限制。飞行时间(Tof)质谱技术的不断发展使得分析能够达到更高的灵敏度、分辨率和质量精度(尤其是亚2 ppm)。Tof MS通常与保留时间偏差、同位素匹配、碎片离子/比率及响应阈值结合使用,用于减少筛查分析中的假阳性和假阴性检测结果。
质谱技术的发展已大大提高了全谱图分析的灵敏度,但进一步提高其灵敏度将能够改善质谱数据的质量。这一点对于保证母离子和碎片离子信息的准确性非常重要,还可确保低分析物浓度水平下的质量精度。尽管MS分析已有如此大的进步,但要快速且高效地鉴定样品中的目标异构化合物仍然极具挑战性(尤其是当样品中含有大量的共提取基质组分时)。已有文献介绍过使用ionKey/MS系统可提升分析的灵敏度,并且具有其它优势5,包括可稀释样品以降低基质抑制效应,从而提高可获得的总体分析物信噪比值。
本应用纪要介绍了碰撞截面(CCS)测量与其它MS新技术联用能够为复杂混合物分析带来的选择性优势。本研究结合了高分辨率质谱与基于离子淌度的高效测量和分离方法。离子淌度是一项快速的正交气相分离技术,能在LC时间范围内获得另一个维度的分离。根据化合物的大小、形状和电荷,可以对化合物进行区分。
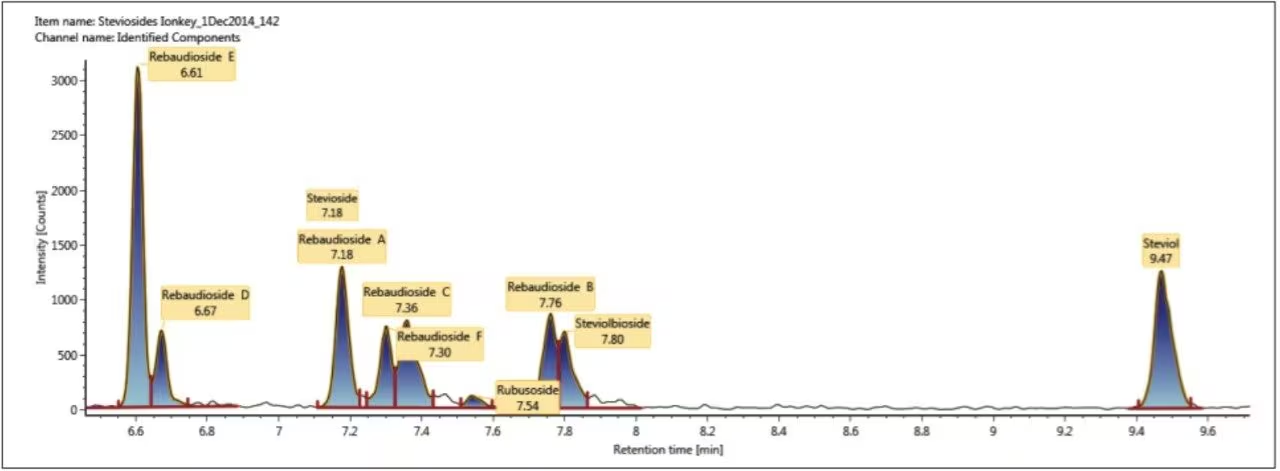
CCS值是离子理化性质中的一个稳定而精确的数值。它是区分每种化合物的化学结构和三维构象差异的一个重要特征6。使用基于氮气的行波碰撞截面(TWCCSN2)测量方法可提高非靶向筛查的特异性。之前得到的TWCCSN2测量结果已录入UNIFI科学数据库。因此我们可利用TWCCSN2的预期值和测定值筛查并确认甜菊糖苷的存在。本文介绍了一种采用ionKey/MS系统和离子淌度质谱(IM-MS)筛查食品中的甜菊糖苷的独特方法,该方法利用了这两项技术明显展现了特异性和灵敏度的优势。目标甜菊醇和甜菊糖苷的结构如图1所示。