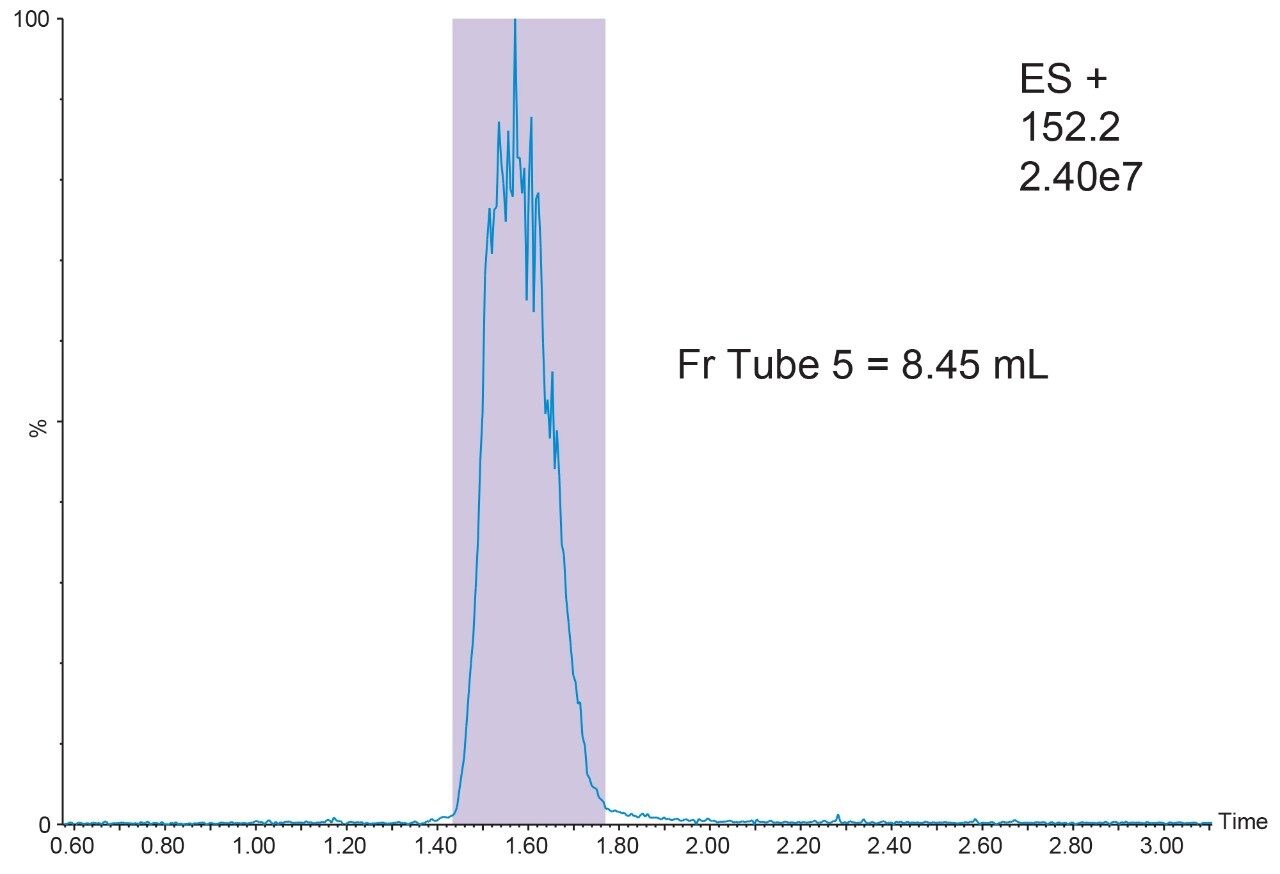
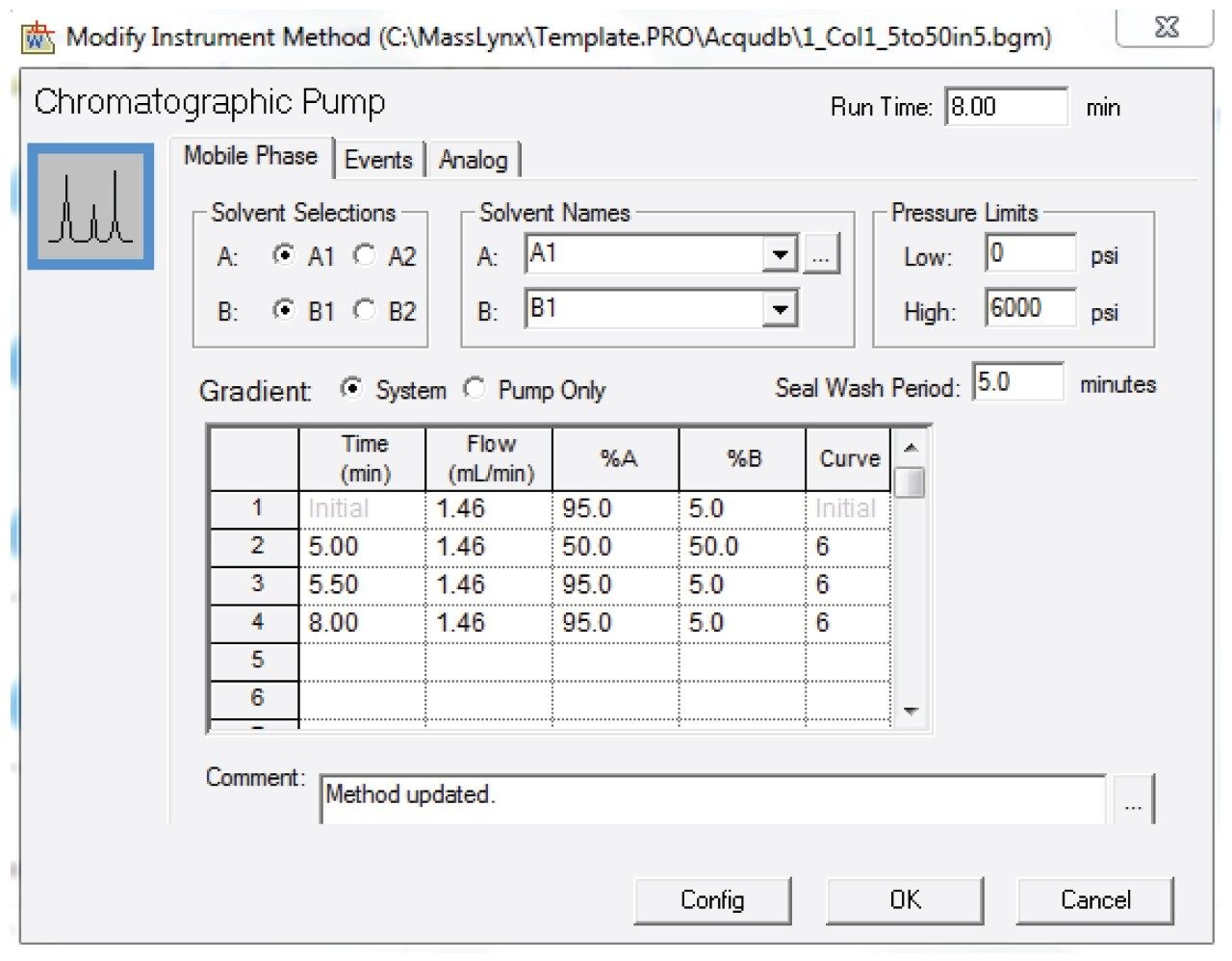
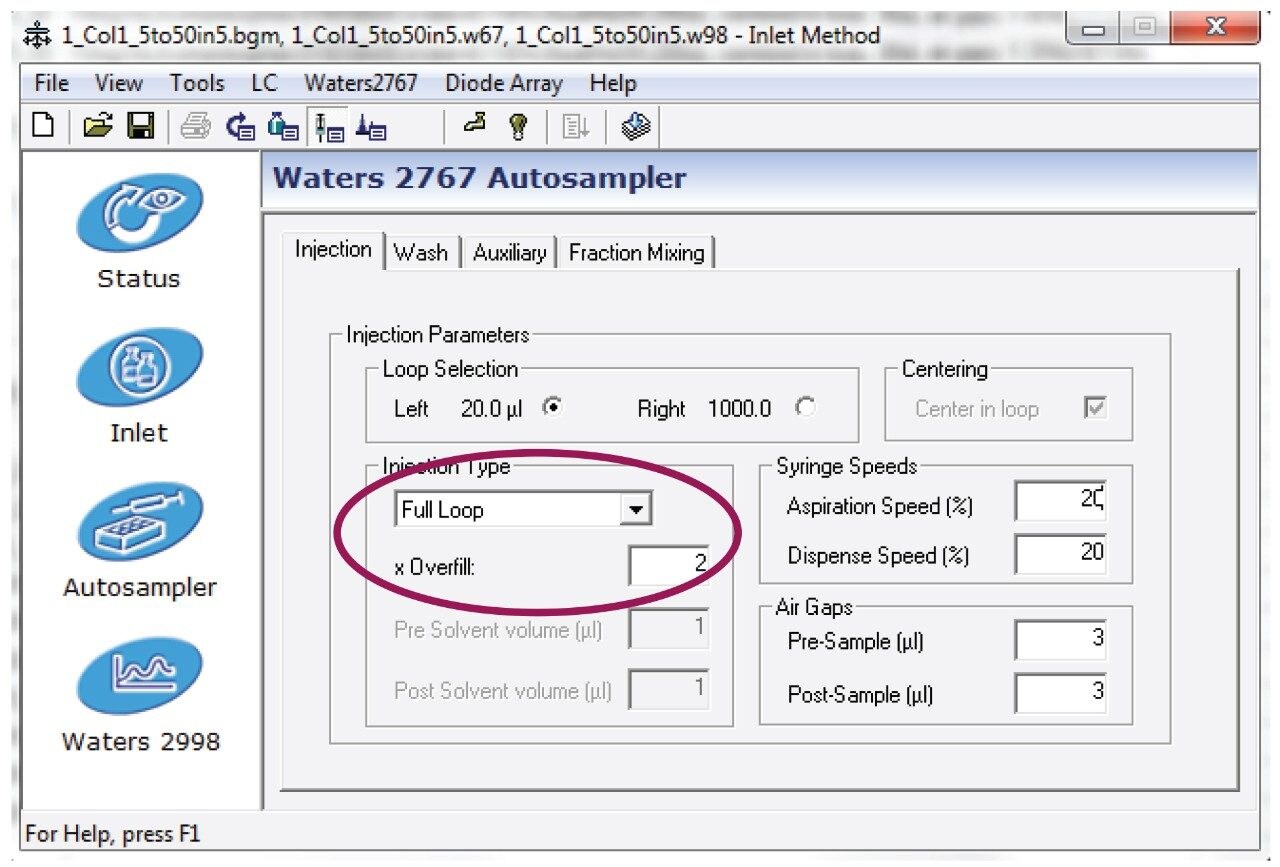
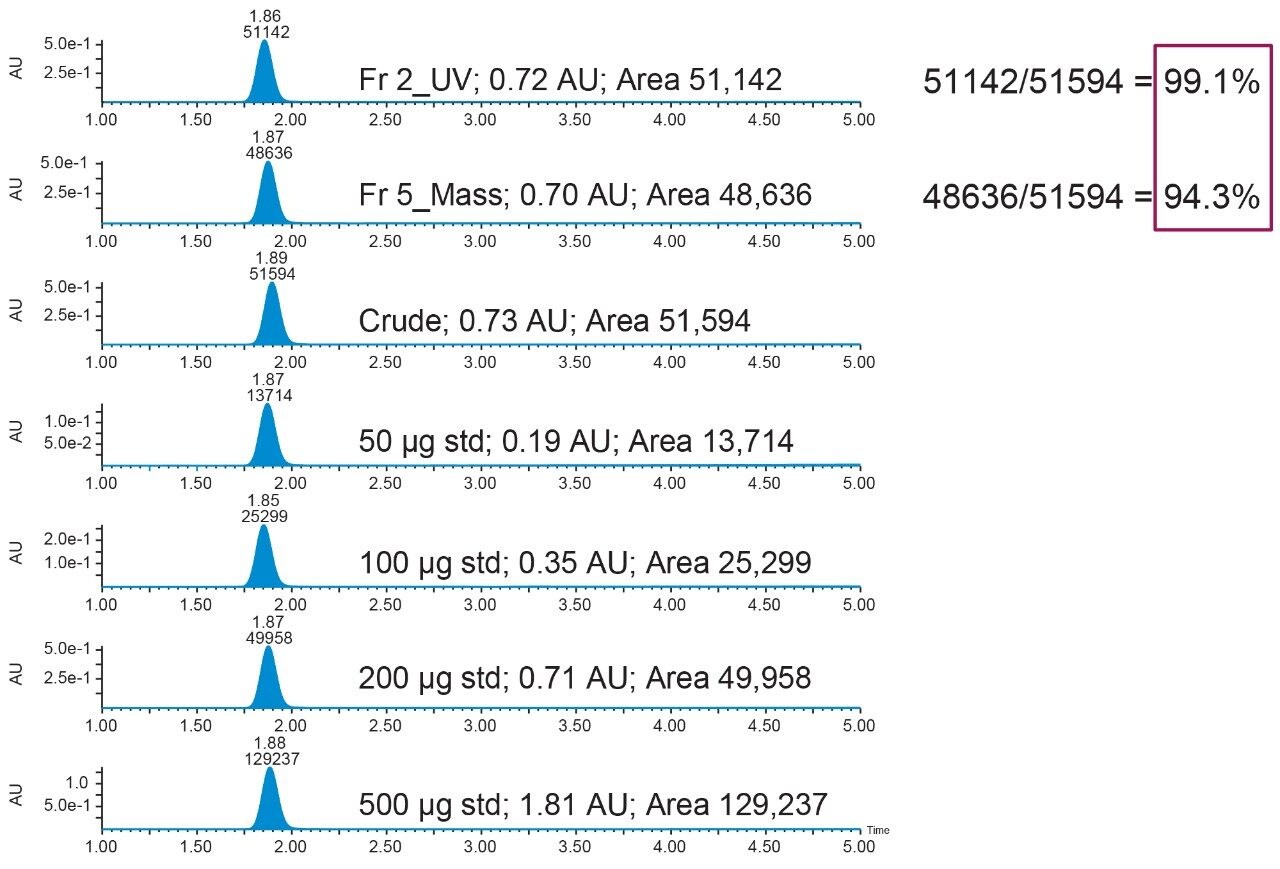
2. Prepared the analyte
A 10 mg/mL acetaminophen stock solution was prepared according to the following protocol.
Note: The exact values noted below were used for demonstration purposes only. The masses used in this procedure do not have to exactly match the ones that are obtained each time this procedure is performed. The values reported here are only meant to be a guideline for the preparation of the acetaminophen stock solution. It is important to note all masses and measures each time the procedure is executed.
A. Weighed approximately 250 mg acetaminophen into a 20 mL scintillation vial. (The mass used in this set of experiments was 250.87 mg.)
B. Prepared 100 mL of 95% water/5% methanol (mixed 5 mL methanol and 95 mL HPLC grade water).
C. Dissolved acetaminophen in ~22 mL of 95% water/5% methanol. Vortexed and sonicated the solution to completely solubilize the analyte.
D. Transferred the whole solution to a 25 mL volumetric flask.
E. Rinsed the vial 3 times with small amounts of diluent and added these rinses to the volumetric flask.
F. Brought the sample volume to 25 mL with additional diluent.
G. Inserted stopper into the volumetric flask and mixed well.
H. Calculated the acetaminophen stock solution concentration:
Acetaminophen concentration = Mass of acetaminophen/volume of diluent
For this example, then: 250.87 mg/25 mL = 10.03 mg/mL