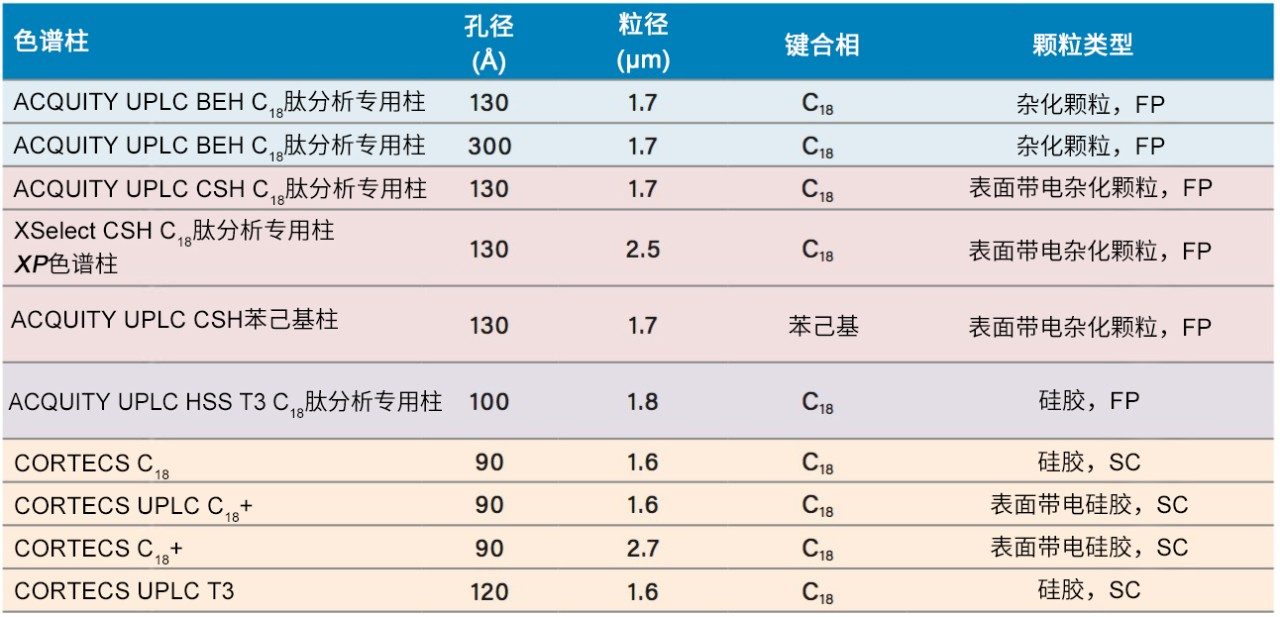
肽选择性注意事项
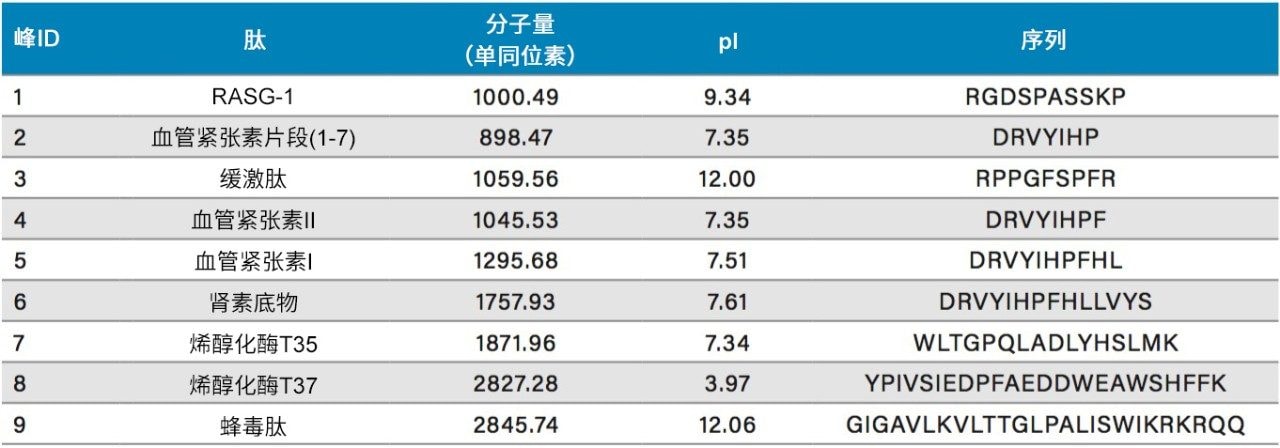
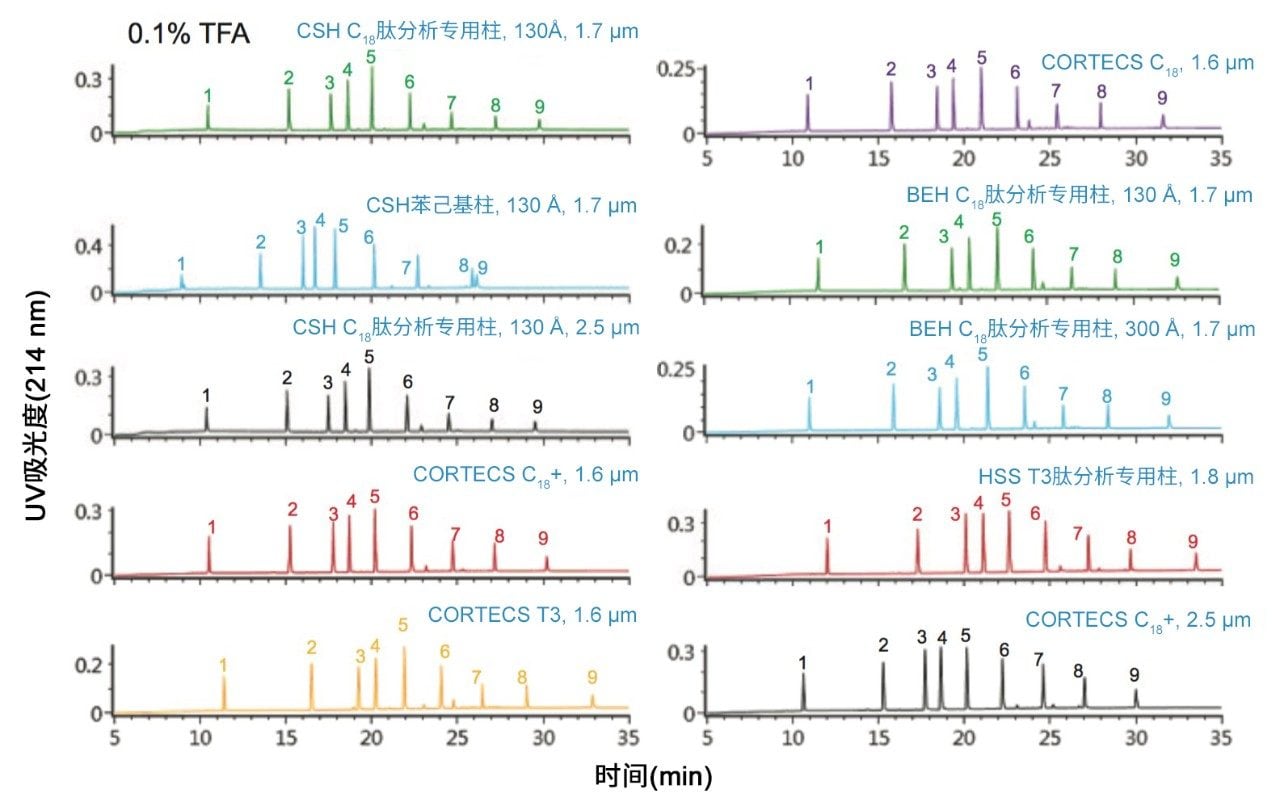
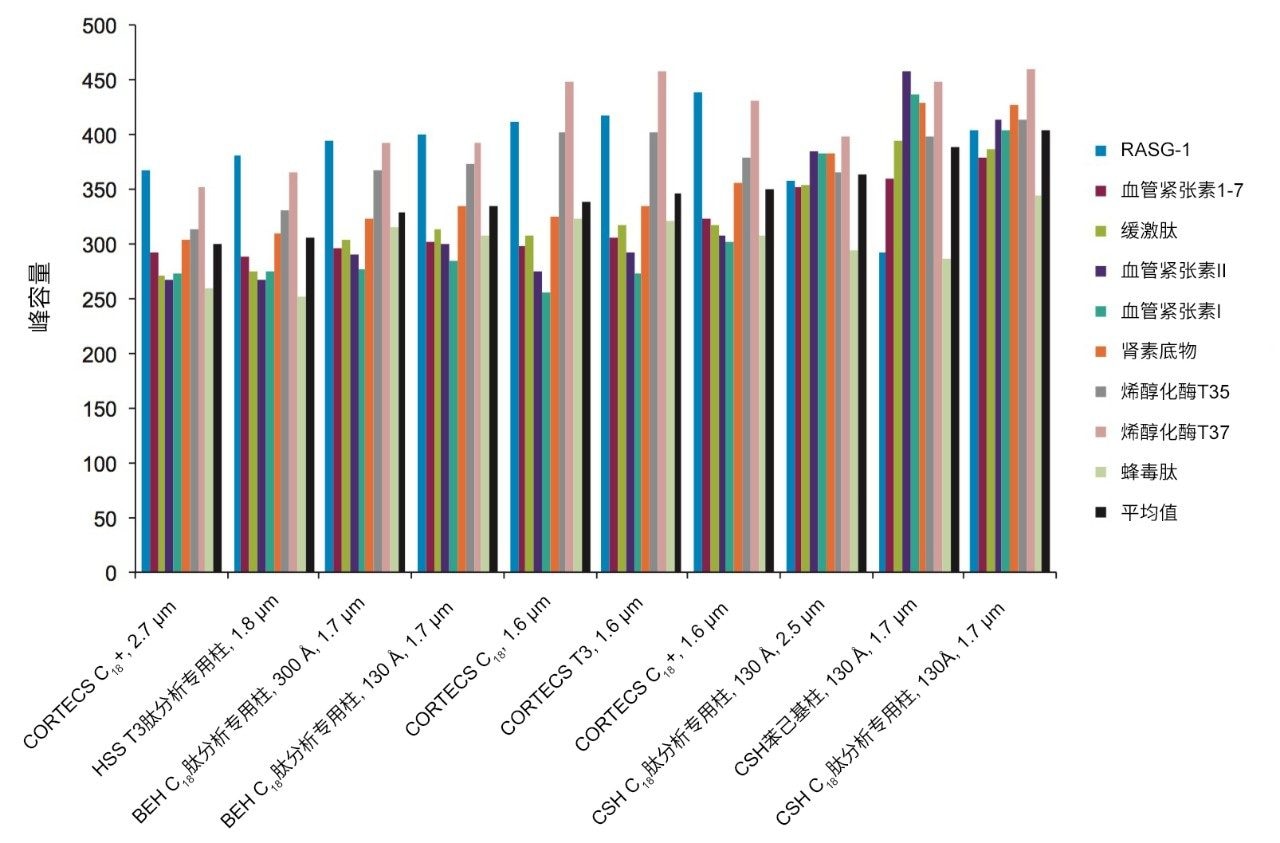
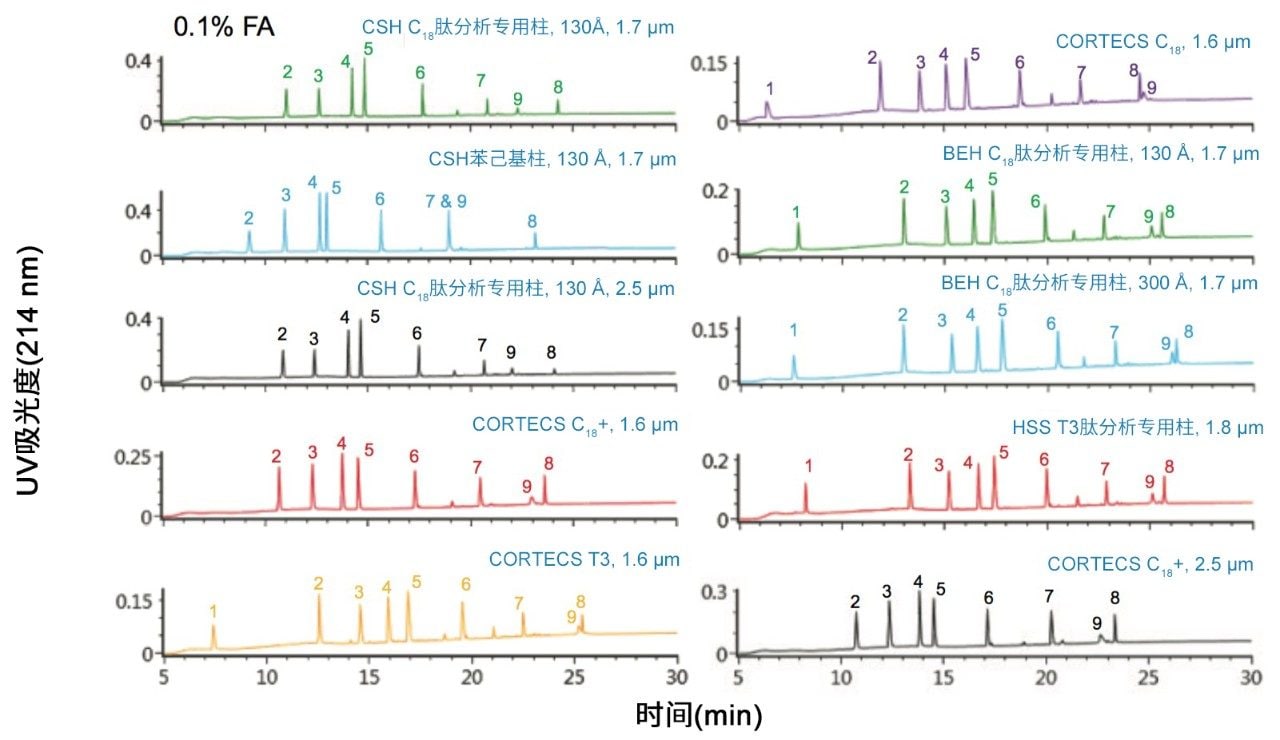
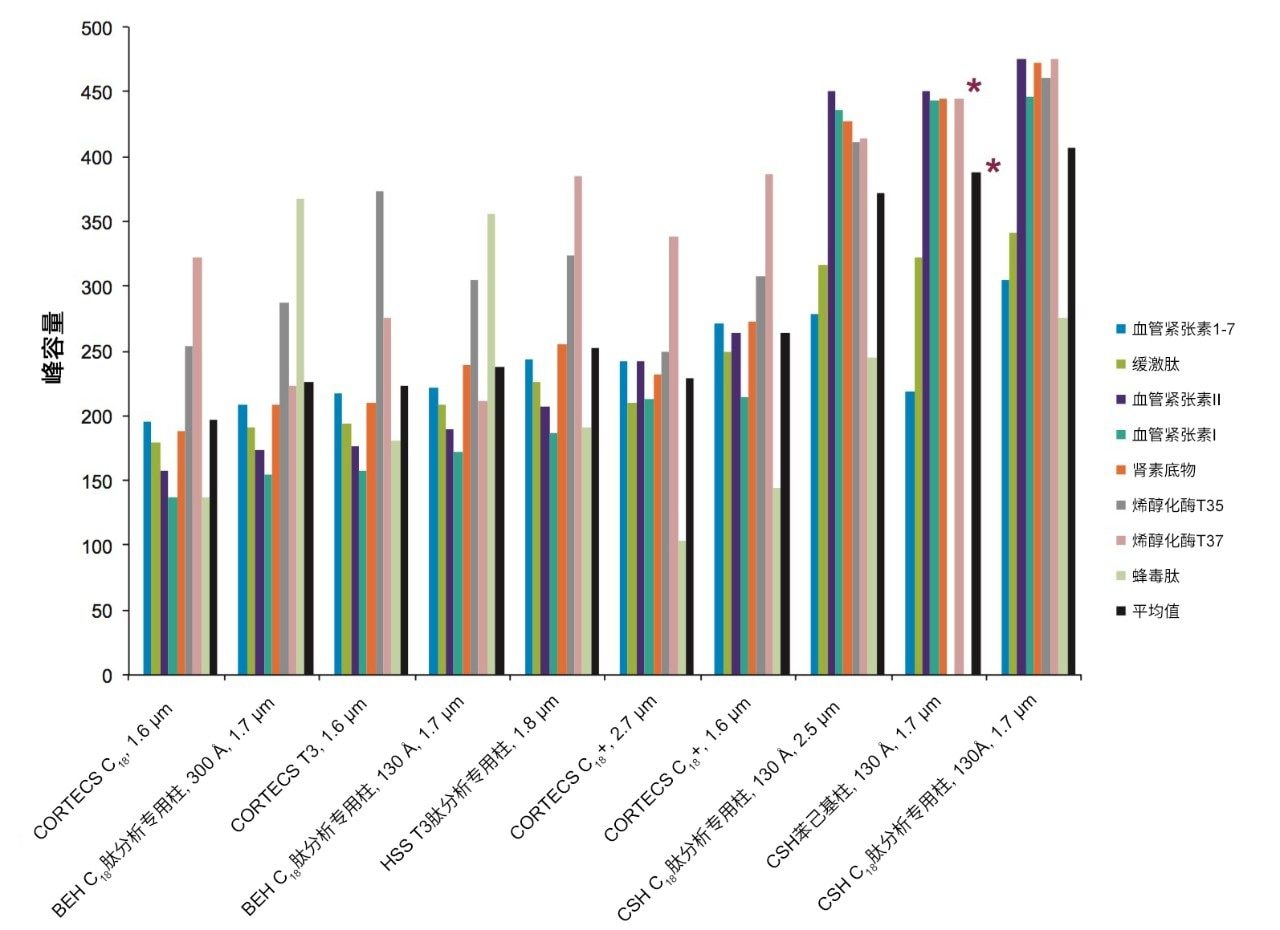
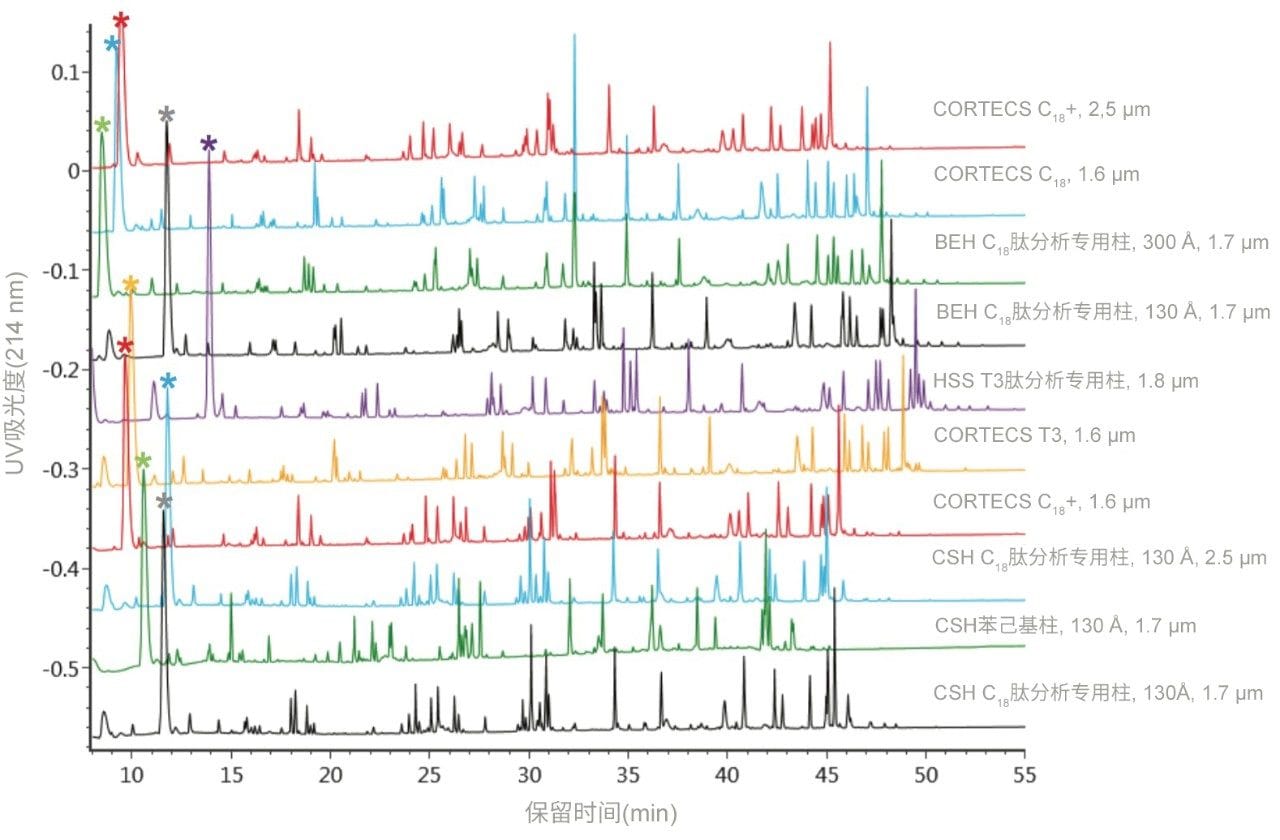
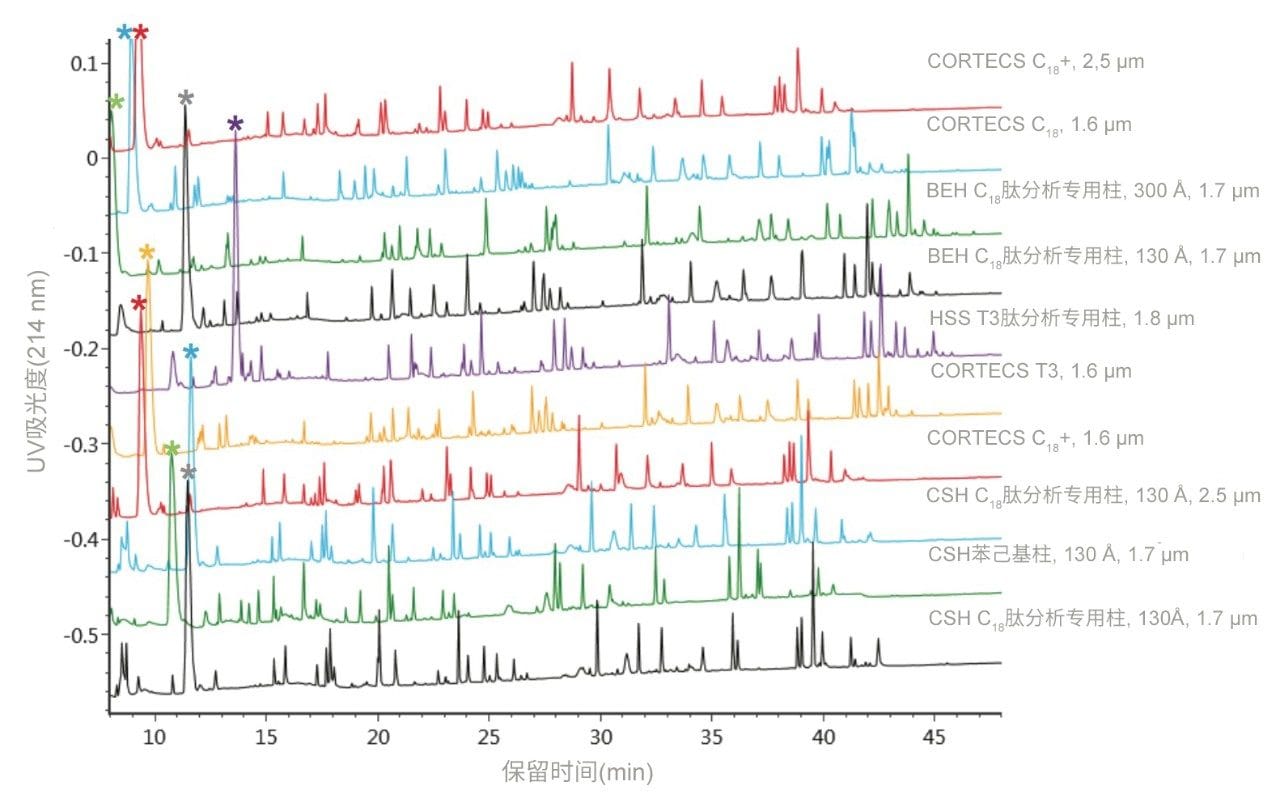
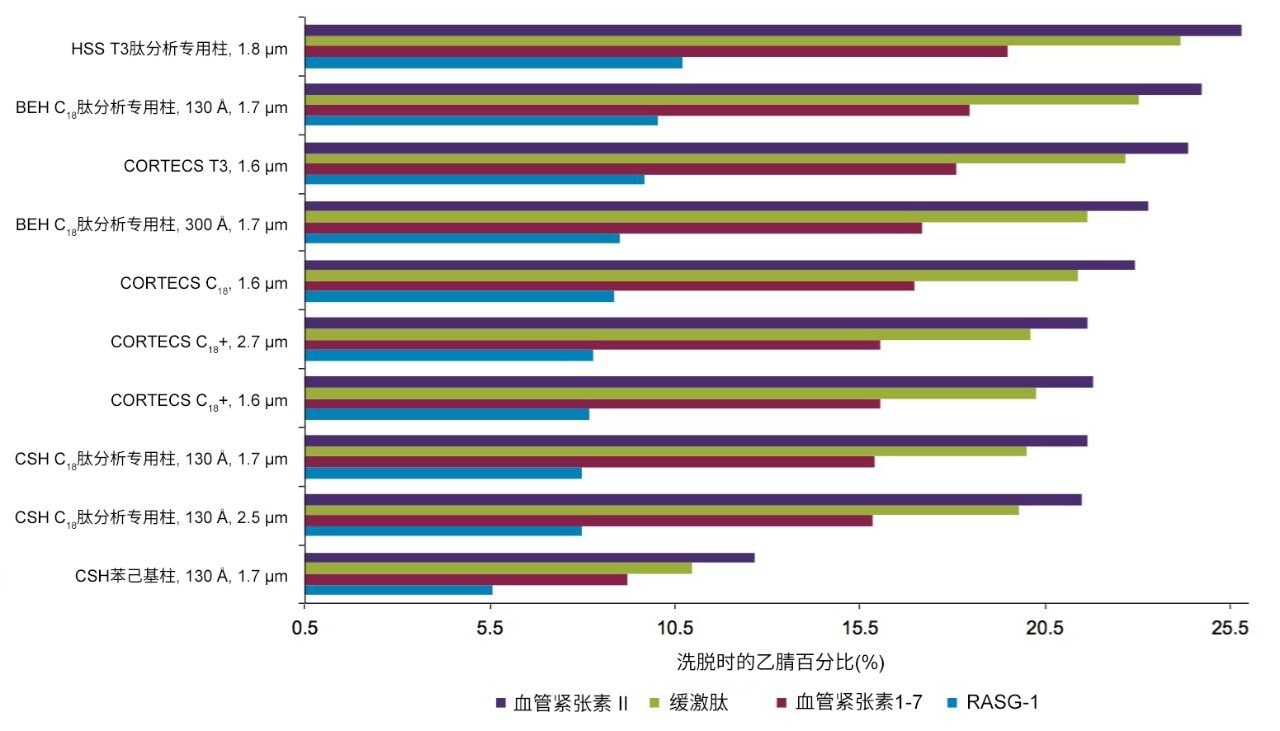
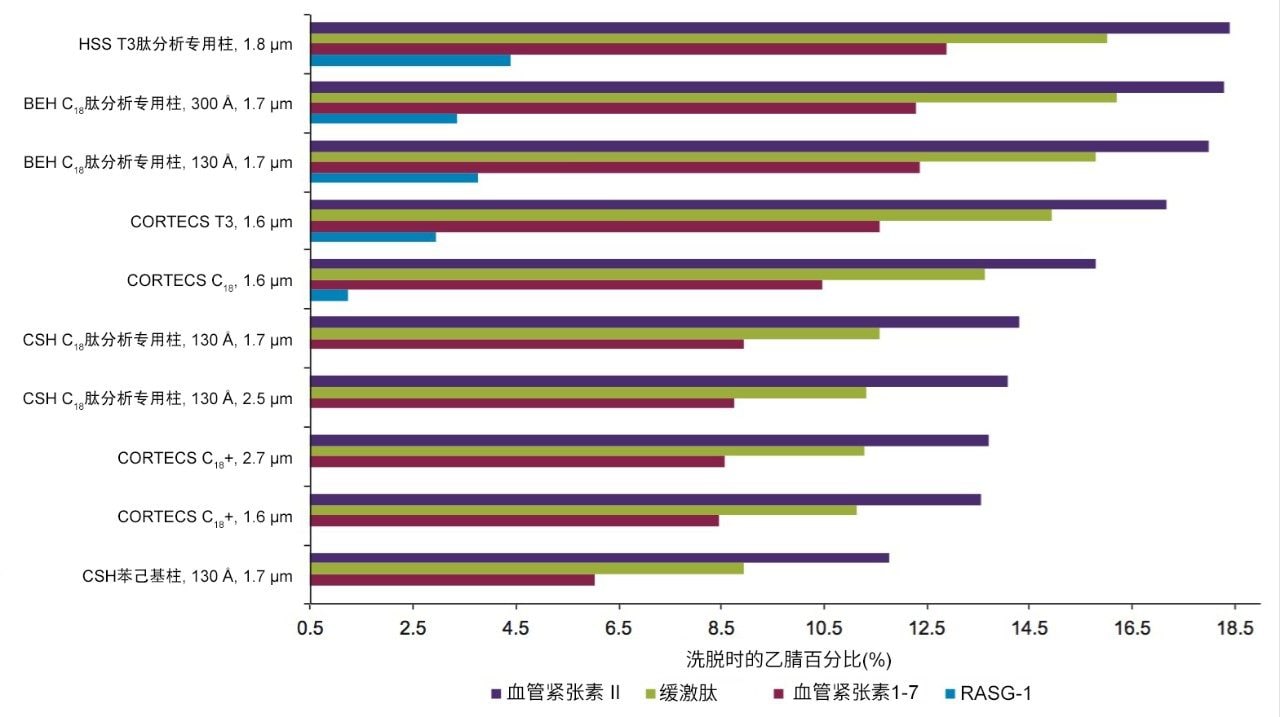
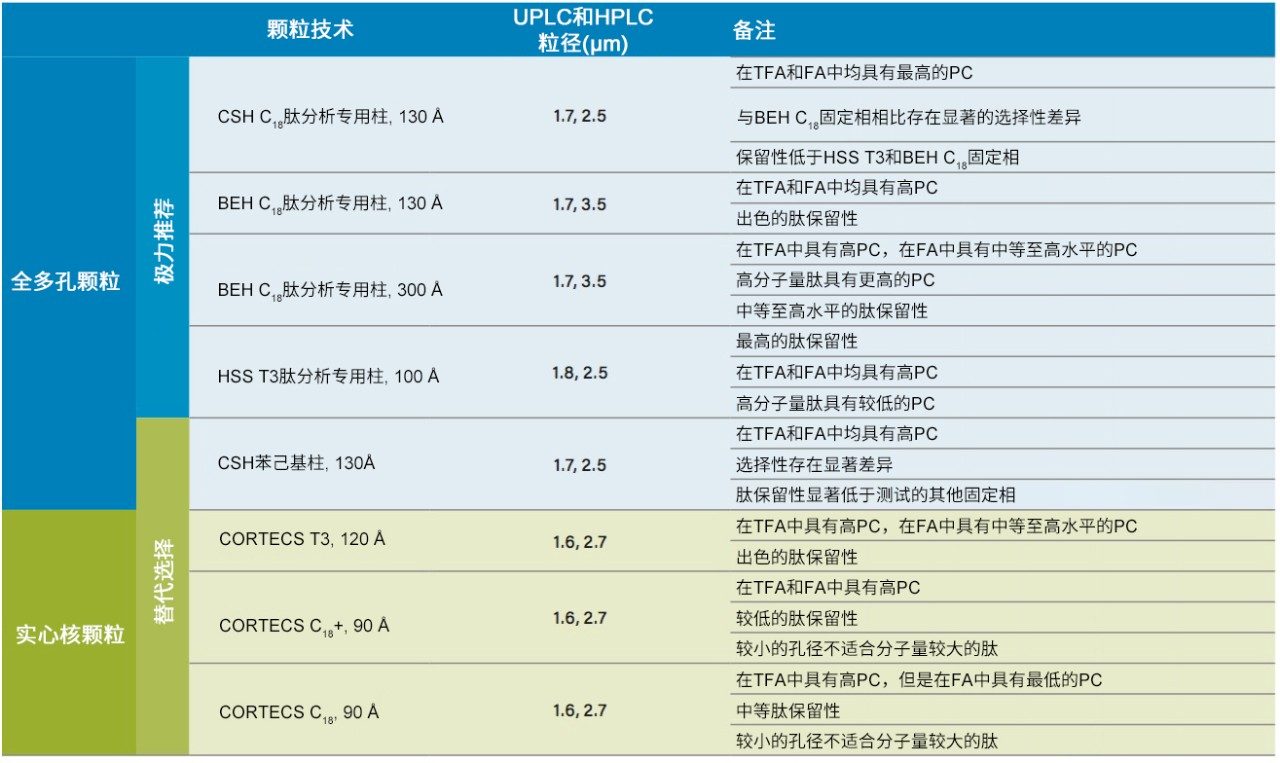
不同固定相之间的选择性差异也可用于改善肽图分析分离方法,即通过调整一对关键的邻近洗脱肽的分离度来实现。但需要注意的是,在这些复杂的肽图分析分离中提高给定的关键肽对的选择性可能导致其他肽对之间的选择性损失。在提供的许多数据中均可观察到选择性差异,这可能是键合相类型、键合相密度、颗粒表面电荷和颗粒孔径等差异的结果。在这些变量中,键合相类型的变化对选择性的影响最显著。例如,图1比较了使用多种C18固定相获得的MassPREP肽混标的分离结果。使用CSH苯己基固定相(130 Å)得到的分离结果显示峰8(烯醇化酶T37)与峰9(蜂毒肽)之间的洗脱顺序发生了变化。烯醇化酶T37在CSH苯己基固定相上的相对保留性更强可能是因为它含有大量芳香族氨基酸残基(酪氨酸、色氨酸和苯丙氨酸)。特别是T37肽中含有的连续苯丙氨酸的氨基酸基序可能与该固定相的苯基键合相形成很强的π-π相互作用。
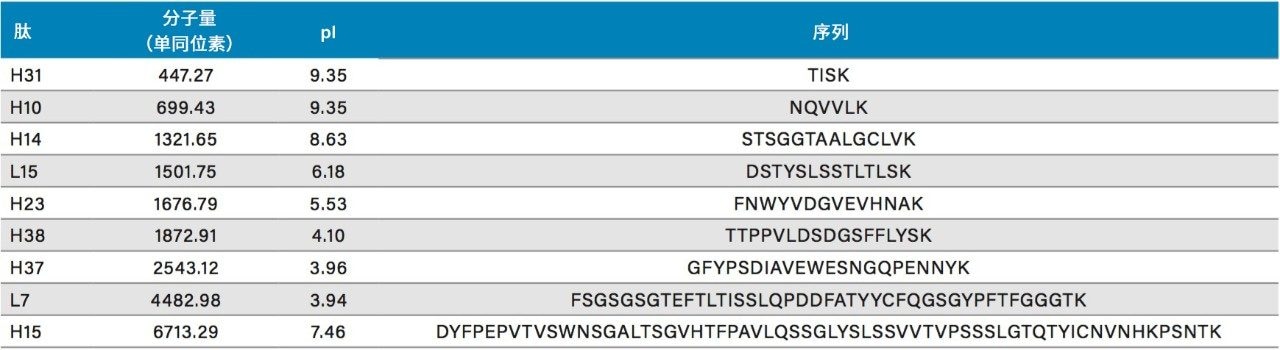
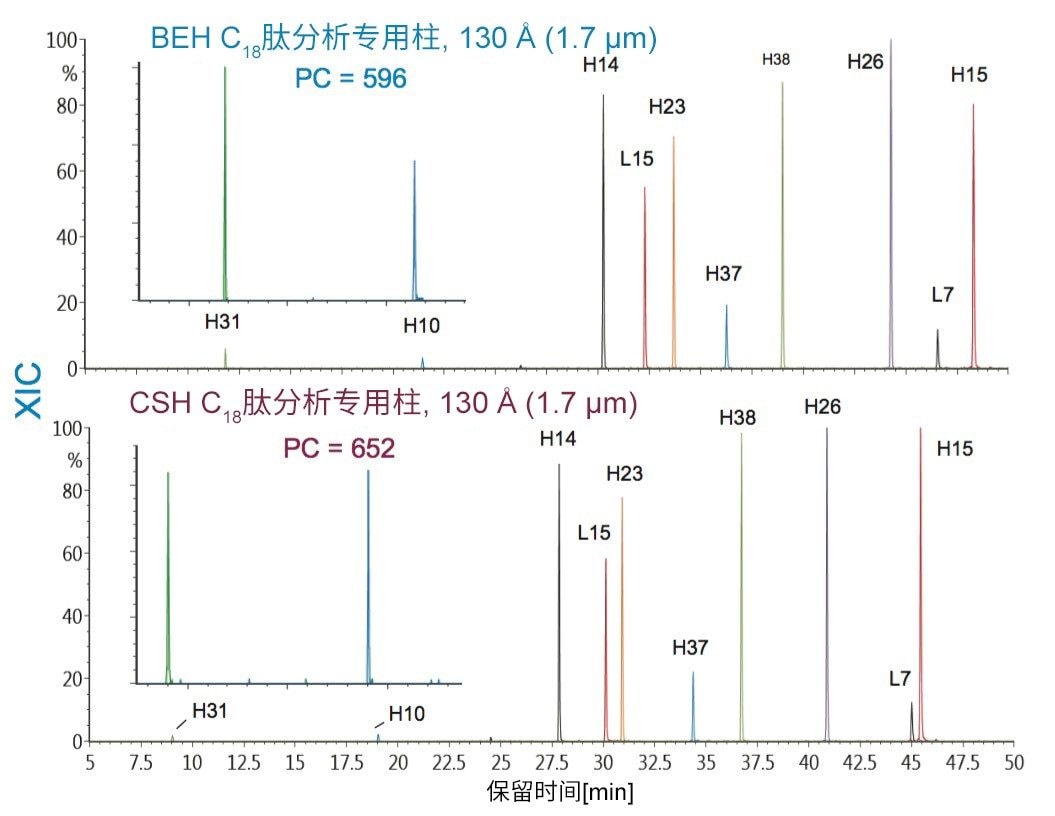
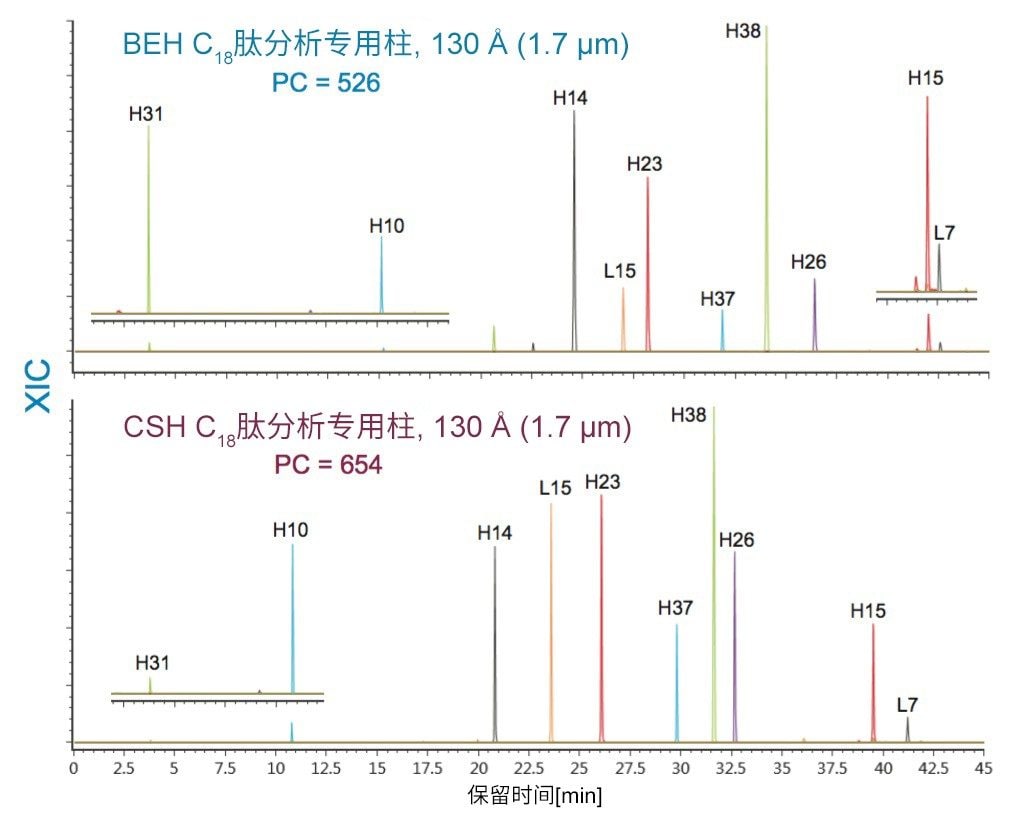
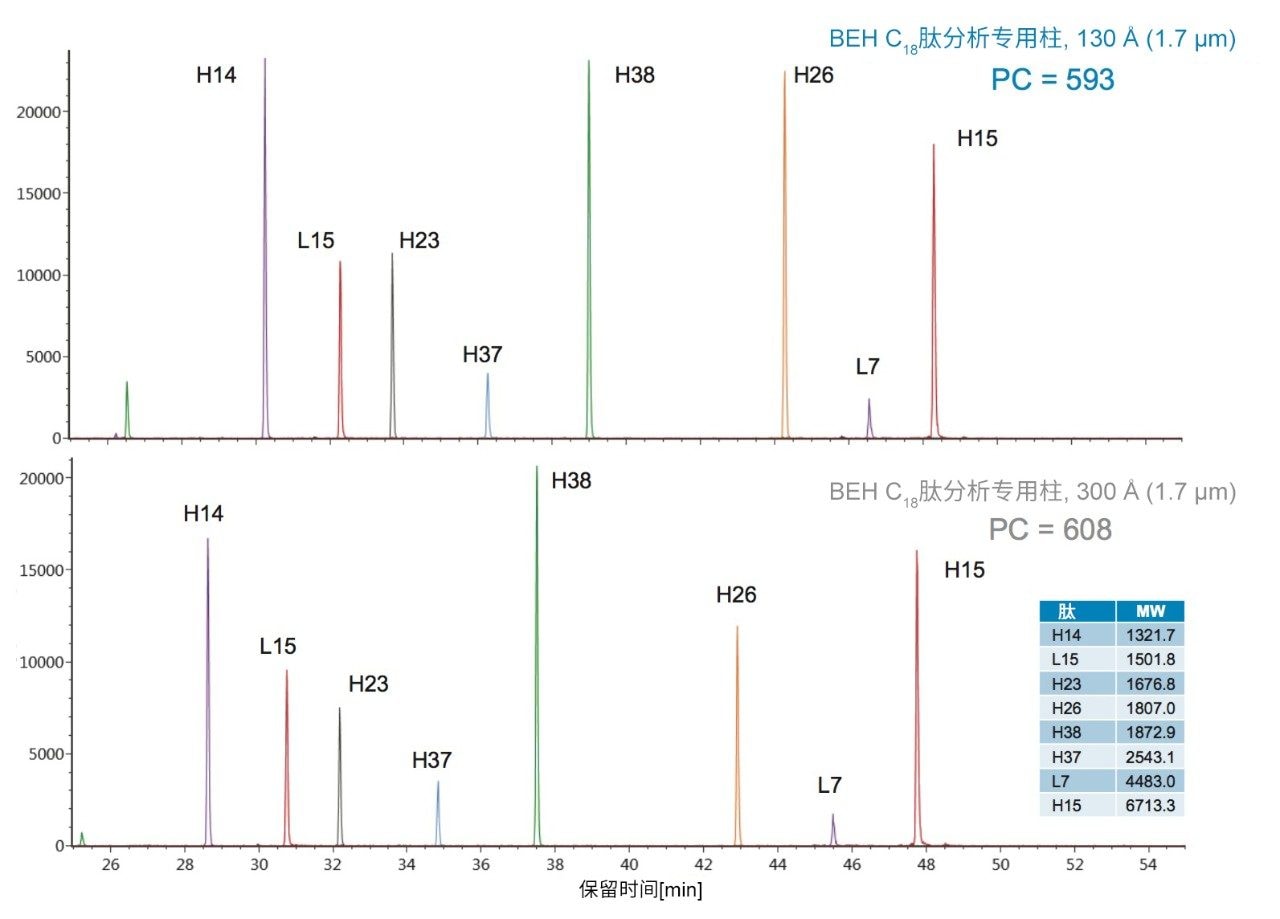
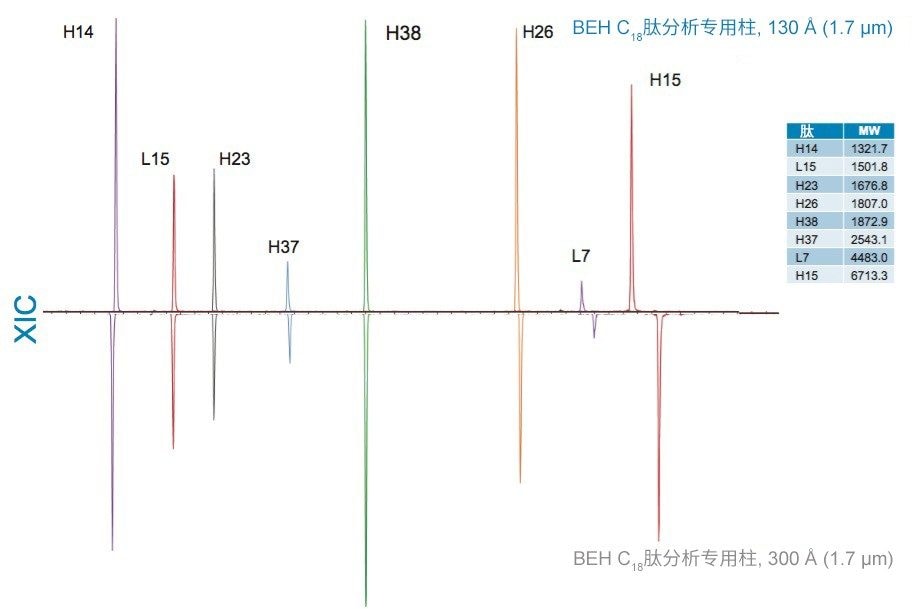
在具有相同键合相的固定相中观察到更细微的选择性差异;但是,除基质颗粒的特性(例如表面电荷和孔径)以外,键合相密度的变化也会影响选择性。这些差异的示例可通过比较还原和烷基化NIST mAb的胰蛋白酶酶解肽图中所选肽的XIC结果观察到。其中,对BEH C18肽分析专用柱(130 Å)与BEH C18肽分析专用柱(300 Å)、CSH C18肽分析专用柱(130 Å)和HSS T3肽分析专用柱(100 Å)进行比较。为更好地显示这些选择性差异,已基于选定肽(H38)对齐色谱图时间轴。
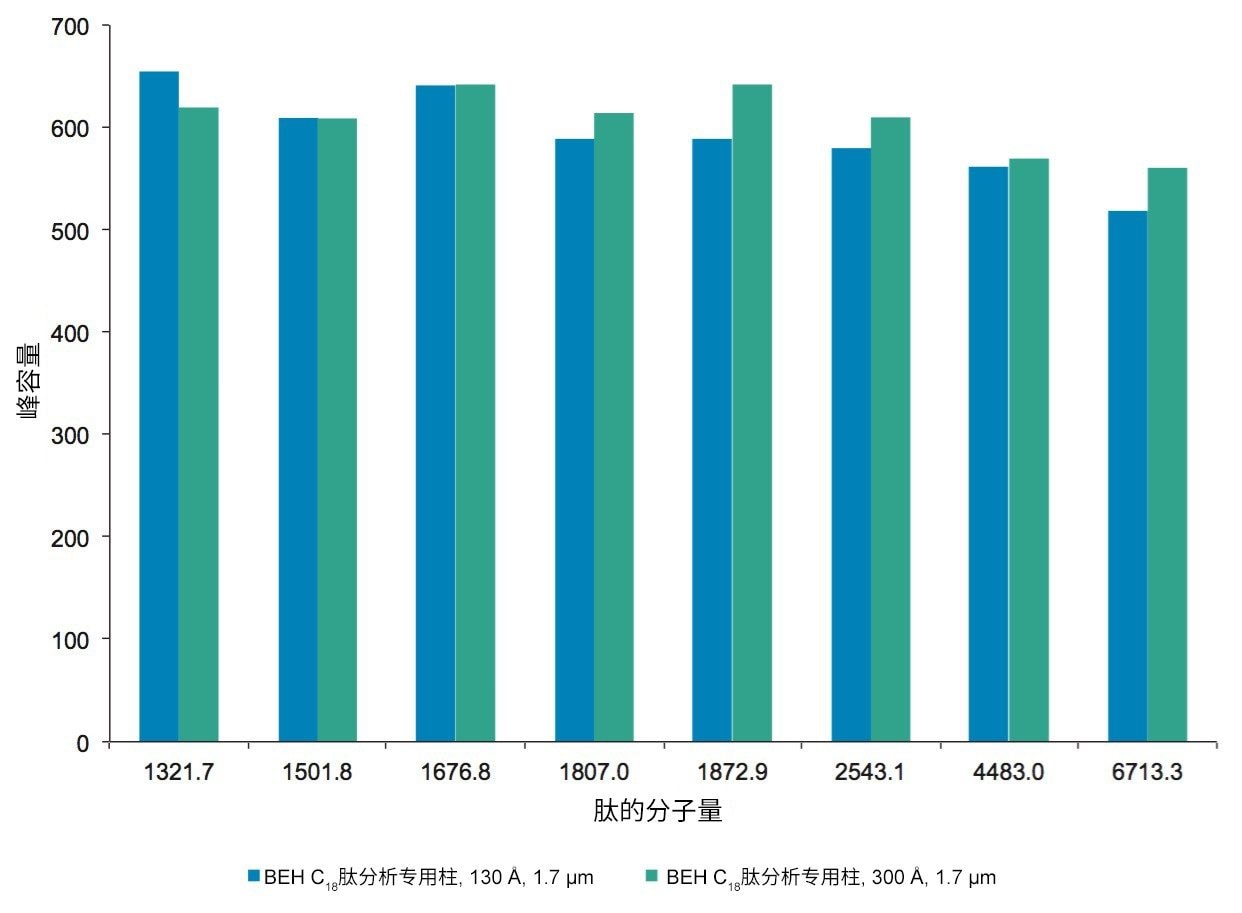
BEH C18肽分析专用柱(130 Å)与BEH C18肽分析专用柱(300 Å)固定相之间的主要区别在于孔径和比表面积(185 m2/g和90 m2/g)。二者的键合相(C18)与键合相密度(3.1 µmol/m2)均相当。因此,随着肽分子大小增加,观察到由体积排阻效应导致的选择性差异。这种效应在两种大分子肽L7和H15中更明显,预计在使用孔径为130 Å的介质时,它们会显著限制分析物进入孔。
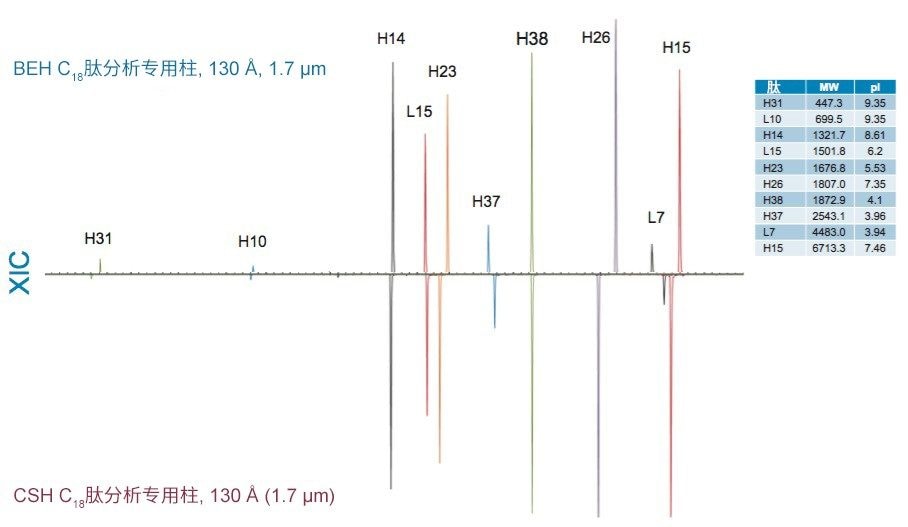
采用BEH C18肽分析专用柱(130 Å, 1.7 µm)得到的分离结果与采用粒径相近的CSH C18肽分析专用柱(130 Å)和HSS T3肽分析专用柱(100 Å)获得的分离结果比较见图14和图15。在BEH C18肽分析专用柱(130 Å)与CSH C18肽分析专用柱(130 Å)之间观察到的选择性差异可能是由于多种因素。这两种颗粒之间的主要区别在于,CSH C18肽分析专用固定相表面施加了正电荷,C18键合相密度也存在细微差异,CSH C18肽分析专用柱固定相的C18键合相密度低约25%(分别为2.3 µmol/m2和3.1 µmol/m2)。值得注意的是,这两种固定相之间显著的表面电荷差异可以在肽(特别是净电荷存在显著差异的肽)的分离中提供潜在有利的选择性差异。
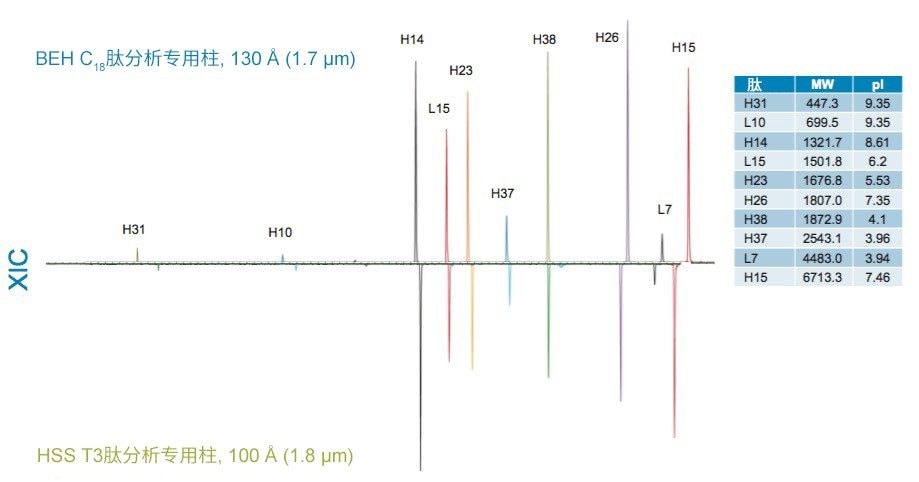
在BEH C18肽分析专用柱(130 Å)和HSS T3肽分析专用柱(100 Å)的比较中,观察到更细微的选择性差异。这些色谱柱在基质颗粒组成(BEH与硅胶)、键合相密度(3.1 µmol/m2和1.6 µmol/m2)、孔径(130 Å和100 Å)和比表面积(185 m2/g和230 m2/g)方面存在显著差异。这种广泛的差异使得难以预测肽选择性变化,因此通常需要进行经验比较,如此处所示。