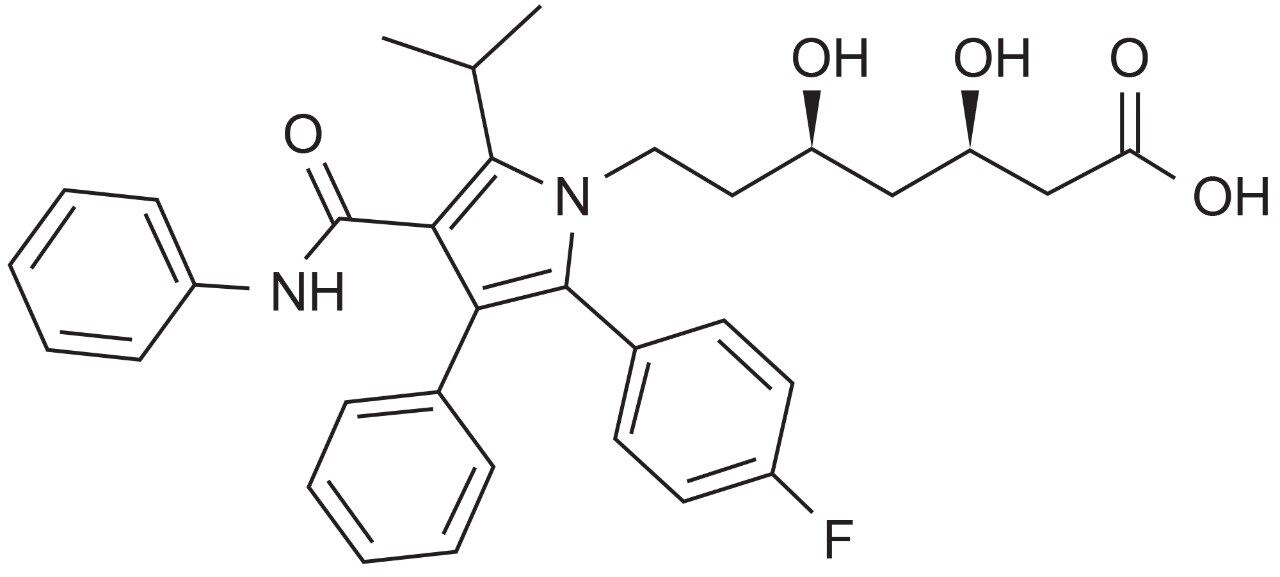
Control of evaporation of mobile phase
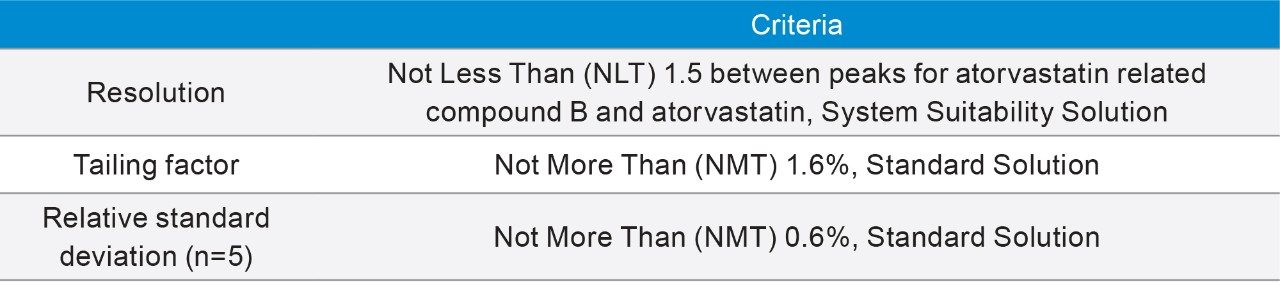
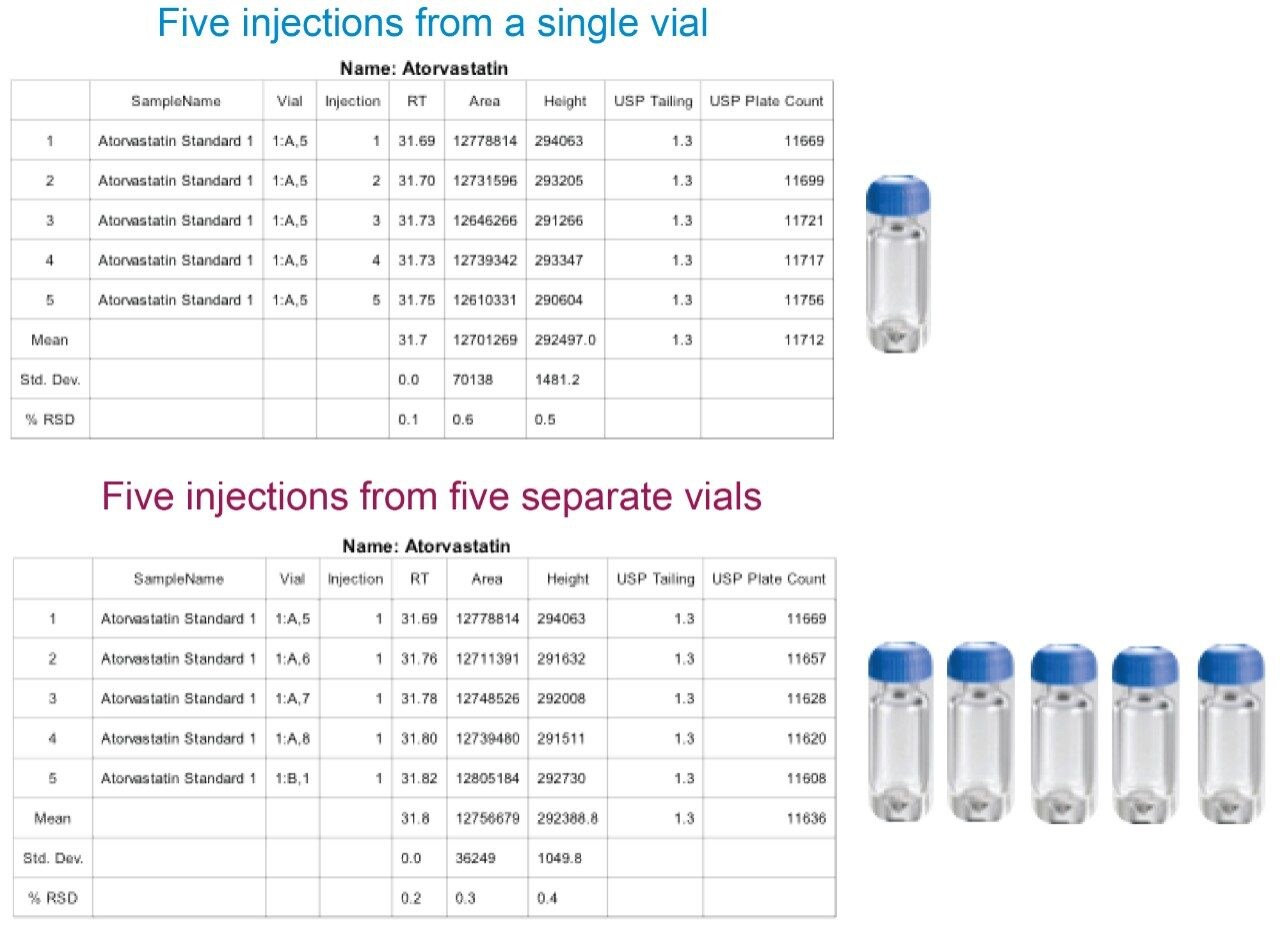
The amount of time to complete testing of an assay can vary depending on number of samples, number of blanks among many variables. With regards to this, USP Chapter <621> on Chromatography describes the number of replicate injections required for a monograph. Specifically “Unless otherwise specified in the individual monograph, data from five replicate injections of the analyte are used to calculate the relative standard deviation, %RSD, if the requirement is 2.0% or less; data from six replicate injections are used if the relative standard deviation requirement is more than 2.0%.”2 Following these recommendations, the USP assay of atorvastatin calcium requires five replicate injections of the standards. However, this is in addition to any blanks, system suitability standards and samples that need to be analyzed. Thus based on the run time (115 minutes), testing of a drug substance requires a minimum of 8 injections (1 blank, 1 system suitability sample, 5 standards, and 1 sample injection) or approximately 16 hours.
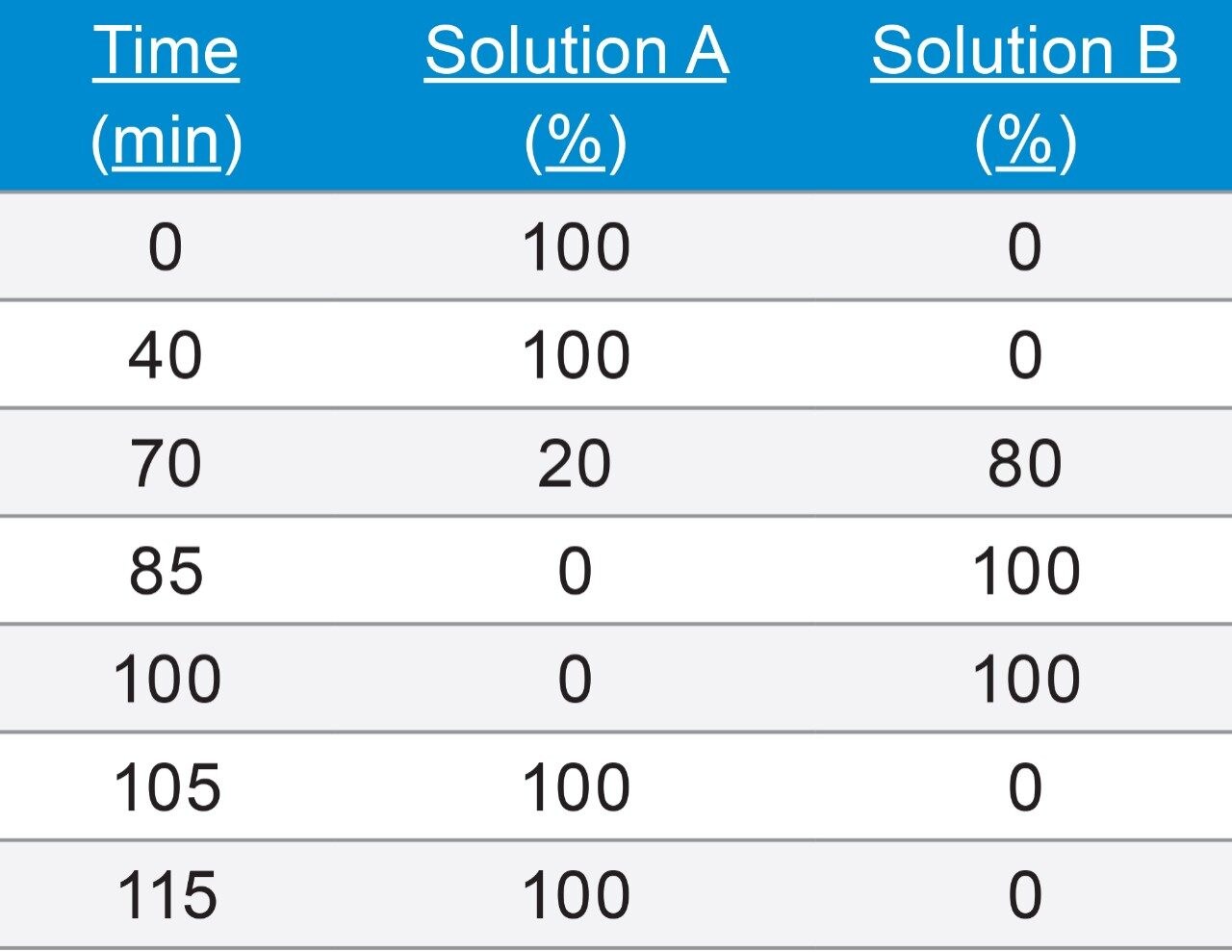
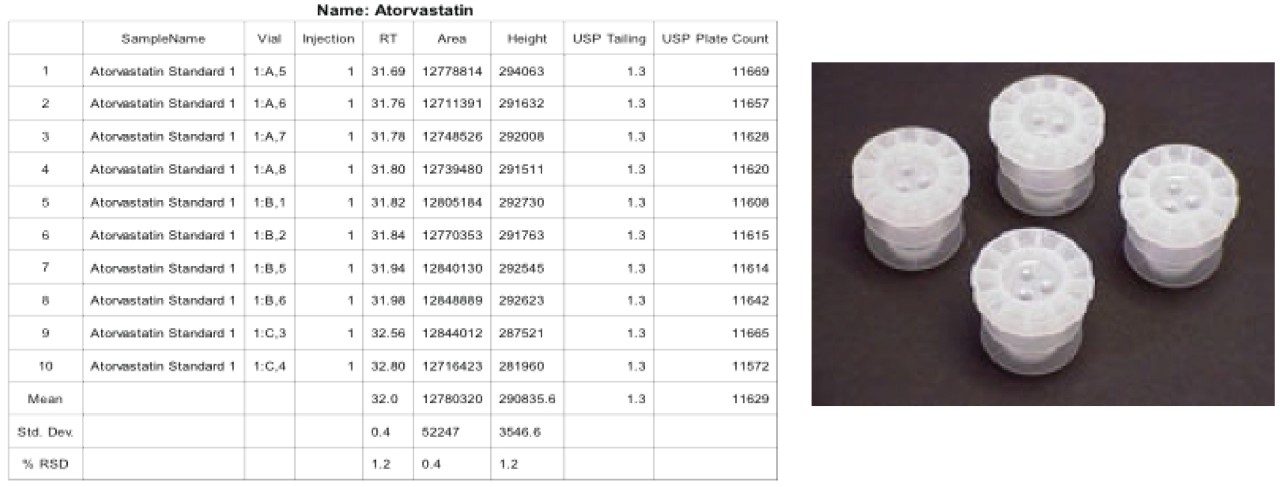
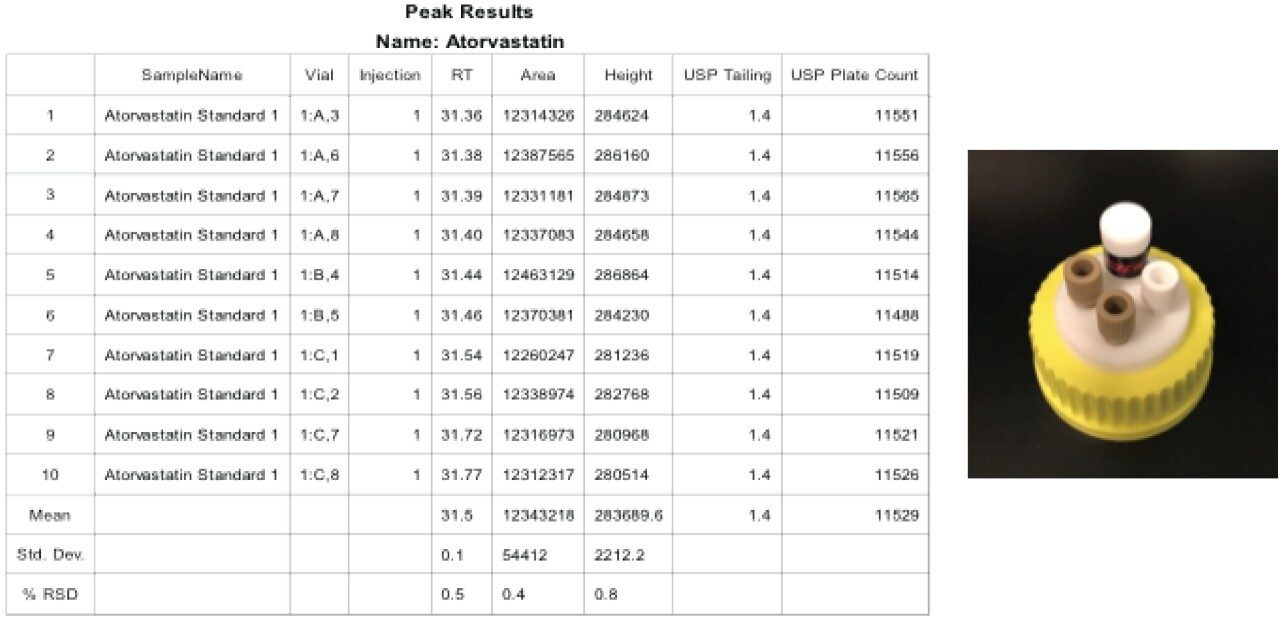
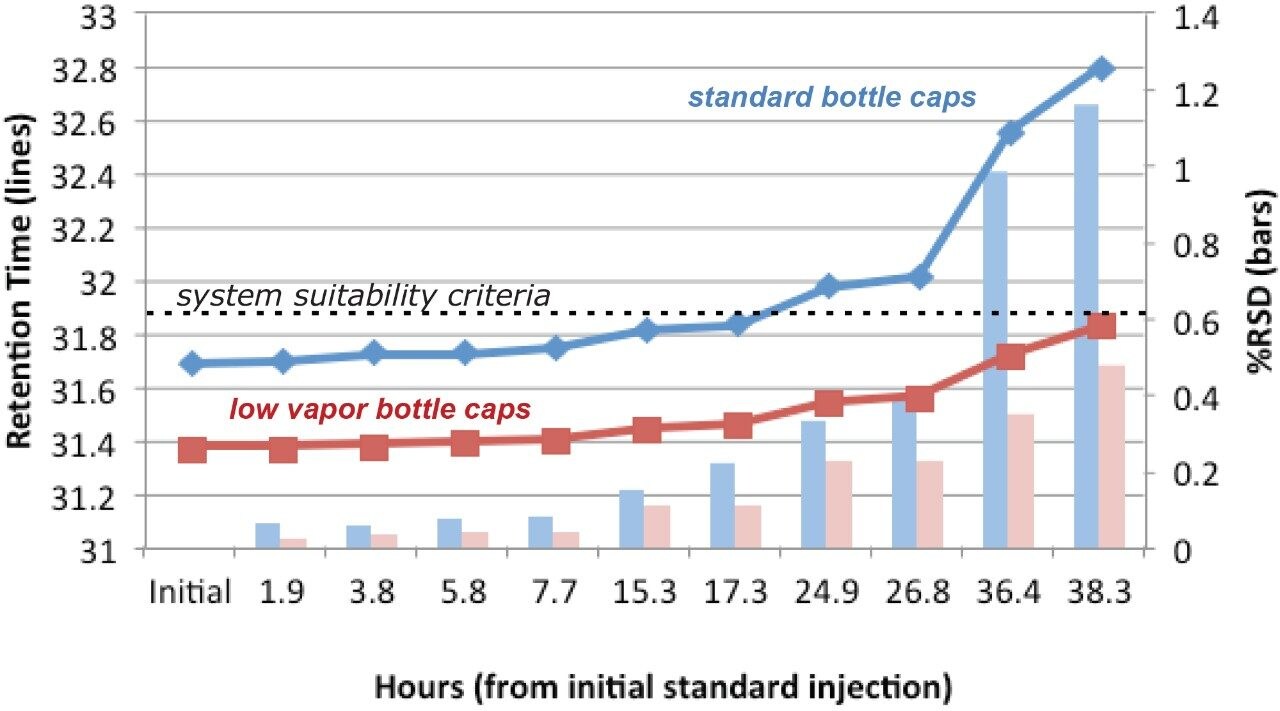
Given the length of the analysis time and the mobile phase components (12% THF in A and B), control of the mobile phase composition is critical for retention time reproducibility. Therefore, the testing was evaluated for a longer period than was required to fully evaluate the mobile phase stability. Specifically the test was conducted over 38 hours and the data was compiled for 10 injections, (blanks and system suitability samples were injected along with standard injections). Standard reservoir or bottle caps were used in this experiment. The results show a significant shift in retention times as well as a retention time RSD of 1.2%, which is outside the system suitability requirement for this analysis (Figure 3). It is likely that evaporation of the organic portion of the mobile phase led to a change in mobile phase composition, a lower “strength” mobile phase with a higher aqueous content thus resulting in the retention time drift.