HILIC可用作在BioAccord系统上进行寡核苷酸完整分子量确认的替代分离模式
摘要
HILIC是一种干净、经济有效的分离方法,易于纳入寡核苷酸完整分子量确认的自动工作流程。如本研究所示,BioAccord LCMS系统能够帮助waters_connect用户对小分子和中等分子寡核苷酸(至多50-mer)执行快速准确的完整分子量确认。
优势
- 自动、合规的HILIC LC-MS工作流程经证明能够提供良好的质量精度(低于15 ppm),适合使用HILIC LC-MS分析进行寡核苷酸完整分子量确认
- 在流动相方面,使用HILIC法分离寡合苷酸与传统的离子对反相(IP-RP)分离相比有三大优势:1)流动相成本至少缩减10倍以上,2)毒性显著减弱以及3) LC-MS操作的流动相稳定性至少提升10倍(可稳定长达2周)
- 与采用不锈钢硬件的传统ACQUITY BEH Amide色谱柱相比,采用MaxPeak高性能表面的ACQUITY Premier BEH Amide色谱柱无需色谱柱老化操作,可节省色谱柱钝化时间,开箱即可直接使用
简介
近年来,寡核苷酸类药物开始作为小分子及治疗性蛋白药物的有力补充1,2。 寡核苷酸类药物的生产和质量控制需要使用高选择性、高灵敏度的LC-MS方法。在寡核苷酸分析中,使用离子对反相色谱法(IP-RP)分离寡核苷酸后进行负离子ESI-MS模式质谱检测是一种广受认可的质谱分析方法。近期有研究使用该寡核苷酸分析方法,介绍了以waters_connect控制BioAccord系统进行数据采集、处理和报告的自动化合规工作流程3,4。图1展示的BioAccord LC-MS系统是沃特世于2019年推出的一套紧凑、稳定、简便易用的平台,可用于生物药物常规分析。本研究使用的这套完全集成的BioAccord LC-MS系统包含一套ACQUITY UPLC I-Class PLUS系统、一台可变波长紫外(TUV)检测器以及ESI-Tof ACQUITY RDa质谱检测器,如图1所示。
 图1.BioAccord LC-MS系统
图1.BioAccord LC-MS系统
在本研究中,我们使用亲水作用色谱法(HILIC)作为IP-RP的替代分离技术,研究了BioAccord LC-MS系统用于寡核苷酸完整分子量确认的能力。近期发表的两篇文章表明5,6,HILIC的普及性虽不如IP-RP色谱法,但它在寡核苷酸分析方面展现出独特潜力。HILIC流动相不采用离子对试剂或有毒且昂贵的挥发性改性剂(例如1,1,1,3,3,3-六氟-异丙醇,HFIP),这是考虑采用此替代分离技术的主要原因7。本应用纪要中的LC-MS数据来自3种不同的化合物:寡核苷酸polyT标准品(OSTs)、经过修饰的寡核苷酸(全硫代磷酸化25-mer寡核苷酸)以及大分子57-mer寡核苷酸。所有数据集均以全扫描MS模式采集,并在waters_connect中使用BayesSpray质谱电荷去卷积算法进行处理,为每种化合物生成准确的完整分子量测量值。
实验
试剂和样品前处理
醋酸铵试剂(LiChropur,产品目录号5.33004.0050)购自Millipore Sigma(美国密苏里州圣路易斯)。乙腈(LC-MS级,产品目录号34881-1L)购自Honeywell(美国卡罗来纳州夏洛特)。HPLC级去离子(DI)水使用MilliQ系统(密理博公司,美国马萨诸塞州贝德福德)净化。使用溶剂A(10 mM醋酸铵的75%乙腈溶液)溶解MassPREP OST(寡核苷酸分离技术)标准品(沃特世部件号:186004135),制得浓度为10 µM的溶液。25-mer全硫代磷酸化寡核苷酸(25-mer PS寡核苷酸序列:5’-C*T*C*T*C* G*C*A*C*C* C*A*T*C*T* C*T*C*T*C* C*T*T*C*T*-3’)、57-mer寡核苷酸(序列:5’-CAA TAT TTT ACA TGA ACT GGA GGT CCG TCA ATG ACA GTG TAG GCT GGA GCT GCT TCG-3’)均购自Integrated DNA Technologies(艾奥瓦州科尔维尔)。两种寡核苷酸均使用溶剂A(10 mM醋酸铵的75%乙腈溶液)溶解,制得浓度为10 µM的溶液。所有寡核苷酸样品的进样量均为2 µL。
LC-MS系统
BioAccord系统,包含ACQUITY UPLC I-Class PLUS系统、TUV检测器和ACQUITY RDa检测器
液相色谱条件
|
色谱柱: |
ACQUITY Premier BEH Amide 1.7 µm, 130 Å, 2.1 x 50 mm,部件号:186009504 |
|
柱温: |
60 °C |
|
流速: |
300 µL/min |
|
流动相: |
溶剂A:10 mM醋酸铵、75%乙腈的去离子水溶液 溶剂B:10 mM醋酸铵、25%乙腈的去离子水溶液 |
|
样品温度:
|
6 °C |
|
样品瓶: |
QuanRecovery MaxPeak样品瓶(部件号:186009186) |
|
进样体积: |
1 µL |
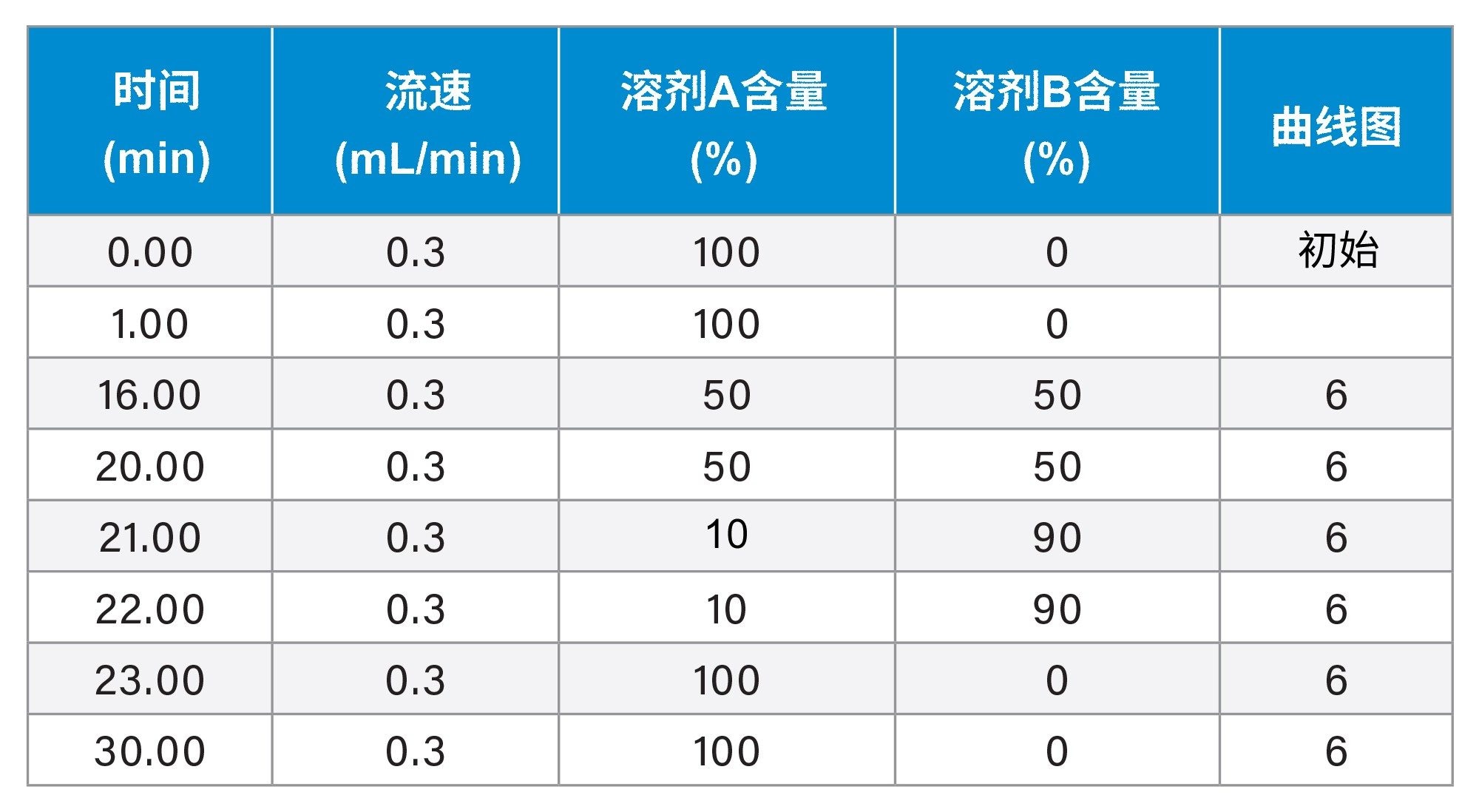
梯度表
清洗溶剂
|
清除溶剂: |
10 mM醋酸铵、25%乙腈的去离子水溶液 |
|
样品管理器清洗溶剂: |
10 mM醋酸铵、25%乙腈 |
|
密封件清洗液: |
20%乙腈的去离子水溶液 |
质谱条件
|
电离模式: |
ESI- |
|
毛细管电压: |
0.8 kV |
|
锥孔电压: |
40 V |
|
离子源温度: |
120 °C |
|
脱溶剂气温度: |
400 °C |
|
脱溶剂气体(N2)压力: |
6.5 bar |
|
Tof质量数范围: |
400–5000 |
|
采集速率: |
2 Hz |
|
实时质量校正标样: |
waters_connect实时校正标准液(部件号:186009298) |
|
数据采集软件: |
waters_connect |
|
数据处理软件: |
waters_connect |
结果与讨论
本研究使用了常规ACQUITY UPLC BEH Amide色谱柱(部件号:186004800)和近期推出的ACQUITY Premier BEH Amide色谱柱(部件号: 186009504)对Waters MassPREP寡核苷酸混标(OST标准品)进行HILIC分离。新色谱柱属于填充亚2 µm颗粒的色谱柱系列,采用了MaxPeak高性能表面(HPS)技术8–12。寡核苷酸含有一个带负电荷的磷酸盐骨架,该骨架可与常规UPLC/HPLC色谱柱中常见的金属表面(如不锈钢外壳或筛板)相互作用。这些相互作用是造成寡核苷酸损失(色谱柱回收率低)、色谱峰形不佳或数据重现性差的常见原因。新安装的色谱柱通常需要通过进样一系列高浓度寡核苷酸样品(柱上>20 pmol)来进行色谱柱钝化,从而减少分析物与流路中金属表面的相互作用。
如图2A所示,使用常规的新ACQUITY BEH Amide色谱柱时,需要进样多达6次才能使OST混合物中所含五种主要寡核苷酸的UV响应值保持稳定。虽然首次进样时的上样量极高(柱上进样20 pmol OST),但保留性明显较差,并且有显著的分析物损失。在后续进样中,分析物和金属表面的相互作用对分离的影响逐渐减少。但是,即使色谱柱经过充分钝化(例如,6次连续柱上进样20 pmol OST),大分子寡核苷酸(dT25、dT30和dT35)的回收率仍然不理想。与此相反,5种主要寡核苷酸在ACQUITY Premier BEH Amide色谱柱上的第1次连续进样与后续进样得到一致的UV响应值,如图2B所示。此外,ACQUITY Premier BEH Amide色谱柱还能够以可重现的方式分离20种微量寡核苷酸杂质(失败序列),适用于dT3至dT24的寡核苷酸。色谱图的重现性反映了UV响应值的稳定性,说明该色谱柱无需色谱柱钝化即可实现分离。MaxPeak HPS层在金属表面和寡核苷酸之间充当屏障,避免寡核苷酸与表面发生螯合,从而显著降低相互作用。本研究表明,ACQUITY Premier BEH Amide色谱柱可实现高分离度寡核苷酸分离,无需色谱柱老化,可直接进样。
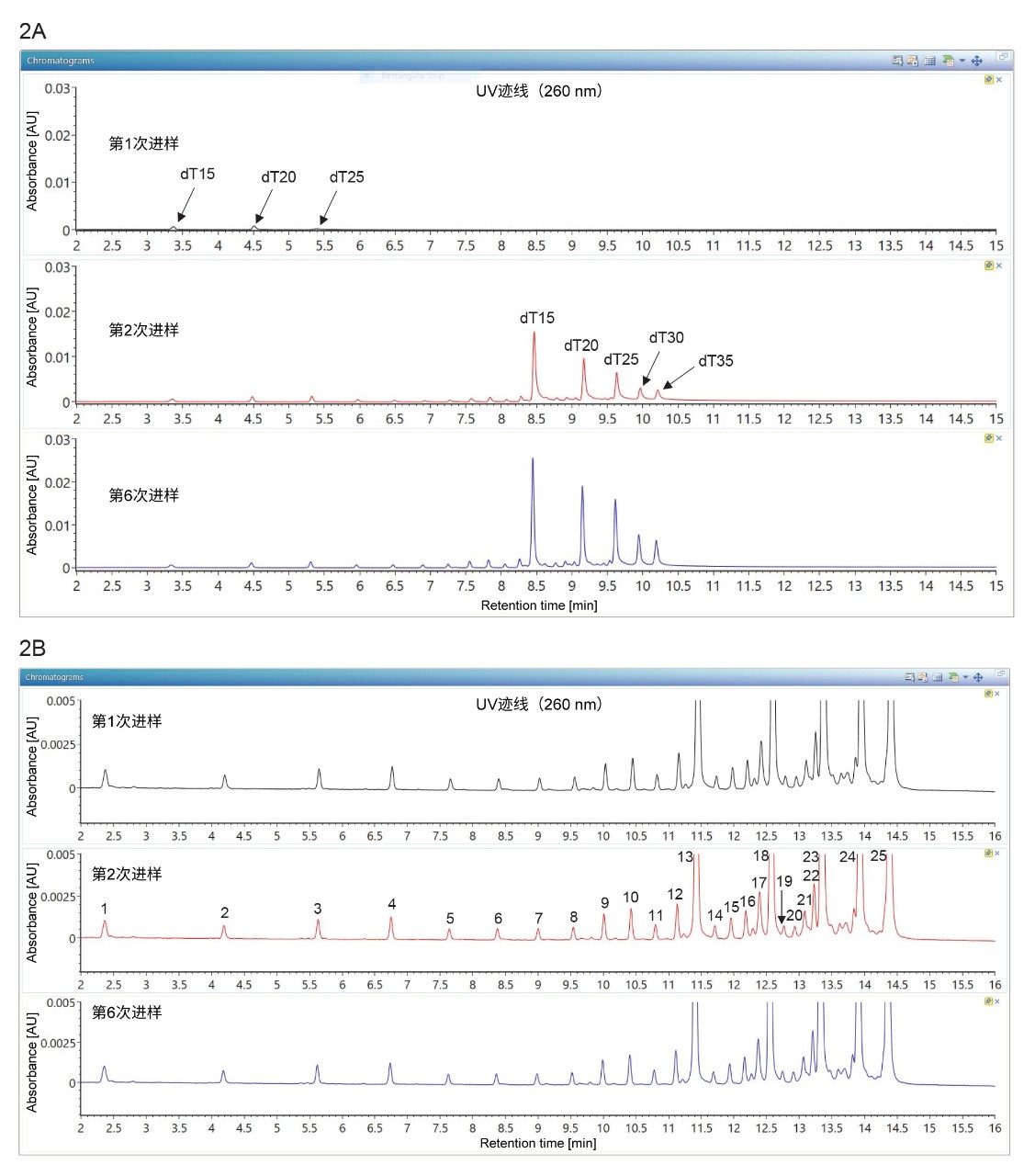 图2.OST混合物在2.1 x 50 mm色谱柱上首次进样的TUV色谱图:(A)常规不锈钢ACQUITY UPLC BEH Amide色谱柱(部件号:1860004800);(B)ACQUITY Premier BEH Amide色谱柱(部件号:186009504)。常规色谱柱需要充分老化后才能得到稳定的UV信号,而ACQUITY Premier色谱柱则不需要任何老化操作。ACQUITY Premier色谱柱的色谱分离重现性良好,无需色谱柱老化/钝化处理,即使用于微量杂质也毫不逊色。图2B中分离的寡核苷酸对应于下列脱氧胸苷失败序列:峰1-dT3、2-dT4、3-dT5、4-dT6、5-dT7、6-dT8、7-dT9、8-dT10、9-dT11、10-dT12、11-dT13、12-dT14、13-dT15(主要成分)、14-dT16、15-dT17、16-dT18、17-dT19、18-dT20(主要成分);根据洗脱顺序,峰19-22分别对应dT21-24,峰23-25分别对应主要成分dT25、dT30和dT35。
图2.OST混合物在2.1 x 50 mm色谱柱上首次进样的TUV色谱图:(A)常规不锈钢ACQUITY UPLC BEH Amide色谱柱(部件号:1860004800);(B)ACQUITY Premier BEH Amide色谱柱(部件号:186009504)。常规色谱柱需要充分老化后才能得到稳定的UV信号,而ACQUITY Premier色谱柱则不需要任何老化操作。ACQUITY Premier色谱柱的色谱分离重现性良好,无需色谱柱老化/钝化处理,即使用于微量杂质也毫不逊色。图2B中分离的寡核苷酸对应于下列脱氧胸苷失败序列:峰1-dT3、2-dT4、3-dT5、4-dT6、5-dT7、6-dT8、7-dT9、8-dT10、9-dT11、10-dT12、11-dT13、12-dT14、13-dT15(主要成分)、14-dT16、15-dT17、16-dT18、17-dT19、18-dT20(主要成分);根据洗脱顺序,峰19-22分别对应dT21-24,峰23-25分别对应主要成分dT25、dT30和dT35。
MassPREP OST标样中5种主要寡核苷酸的HILIC ESI-MS谱图见图3A。相同化合物的IP-RP ESI-MS谱图表现出不同电荷态的双峰分布3,而HILIC ESI-MS谱图的电荷明显更少,每种寡核苷酸可见3种主要的高丰度电荷态。IP-RP色谱分离依赖于带正电荷的烷基胺(溶解于高pH值流动相(pH 8–10),以形成强离子对)和带负电荷的寡核苷酸磷酸盐骨架。这些非共价相互作用不仅对寡核苷酸分离有益且不可或缺(因为烷基胺持续与RP色谱柱的C18疏水链相互作用),还会影响寡核苷酸结构,如IP-RP色谱法得到的ESI-MS寡核苷酸谱图中的双峰分布所示3。IP-RP色谱法使OST寡核苷酸部分变性,产生两种电荷态分布:一种包含低电荷态(-3至-5),对应于天然构象;另一种变性的寡核苷酸构象包含高电荷态(-7至-15)。HILIC分离不需要使用离子对试剂,寡核苷酸保留基于流动相和富水层之间的分配机制,并且部分固定于固定相上13–14。如图3A中的ESI-MS谱图所示,HILIC相互作用似乎可保留OST的天然状态构象,其中包含数量有限的电荷态(仅3种),所有检出结果均在相对高的质量数范围内(m/z = 1000–3000)。对这些大幅精简的ESI-MS谱图(与IP-RP谱图常见的7-12电荷态谱图相对比)进行BayesSpray去卷积后得到了非常精确的完整分子量测定值,如图3B的处理结果所示。所有主要OST的质量精度均优于15 ppm,与此前在IP-RP分离中观察到的结果相同3。图3A中的HILIC ESI-MS谱图包含极高水平的钠和钾加合物离子(裸寡核苷酸信号MS响应值的20%~40%),这表明天然寡核苷酸与流动相中的痕量金属之间可能存在紧密关联。因此,HILIC LC-MS分析的灵敏度弱于变性的IP-RP LC-MS分析。
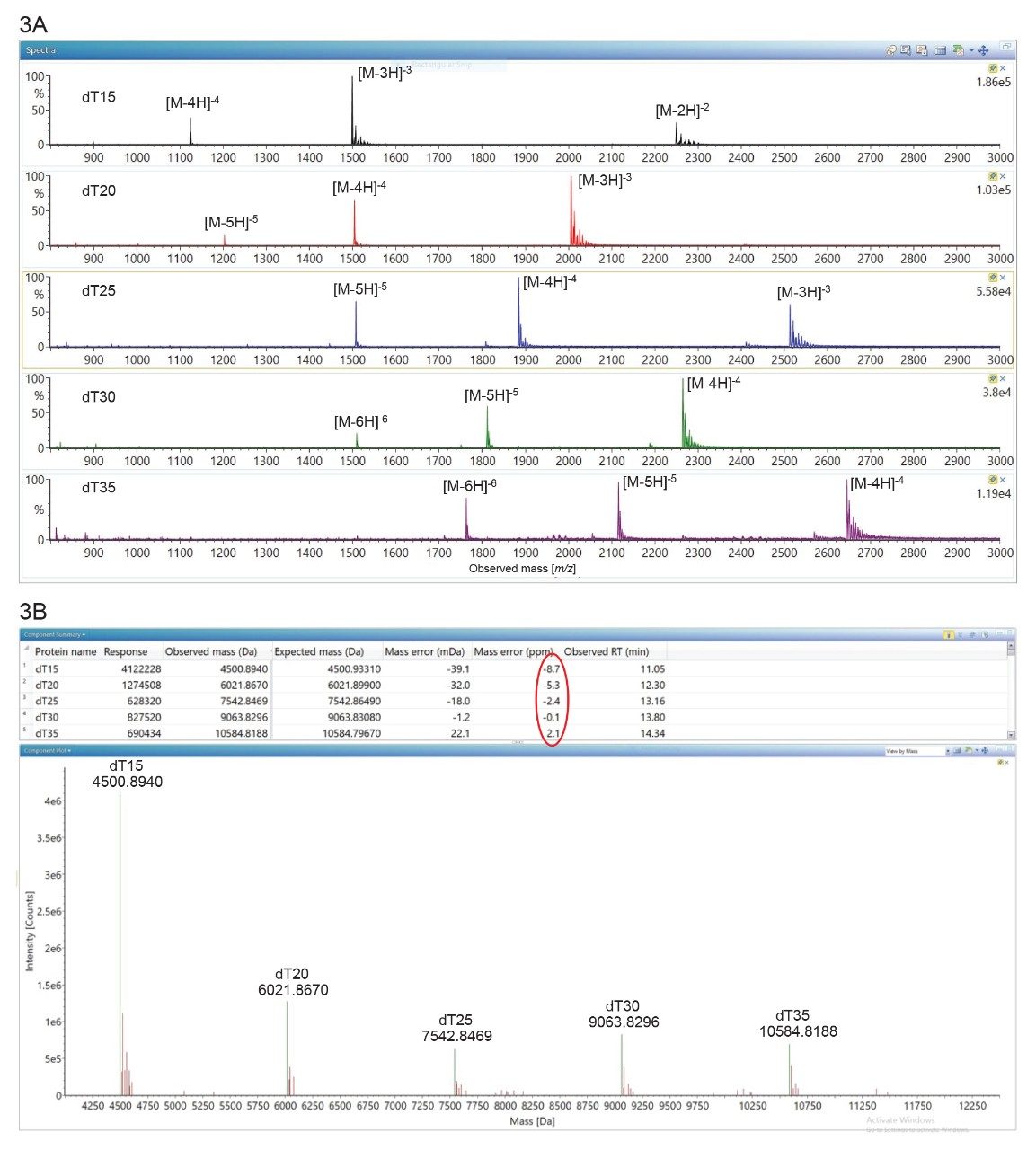 图3.(A) OST混合物中5种成分的HILIC ESI-MS谱图。在所有寡核苷酸中仅检出3种高丰度电荷态,表明OST在气相条件下呈天然状态构象;(B)在waters_connect软件中对OST谱图进行BayesSpray处理后得到的电荷去卷积OST谱图。五种化合物的质量精度均优于15 ppm。
图3.(A) OST混合物中5种成分的HILIC ESI-MS谱图。在所有寡核苷酸中仅检出3种高丰度电荷态,表明OST在气相条件下呈天然状态构象;(B)在waters_connect软件中对OST谱图进行BayesSpray处理后得到的电荷去卷积OST谱图。五种化合物的质量精度均优于15 ppm。
25-mer全硫代磷酸化寡核苷酸(25-mer PS oligo)是1型人体免疫缺陷病毒(HIV-1)的一种候选治疗药物15,16,本研究通过HILIC LC-MS方法,使用与MassPREP OST混标相同的实验条件分析了该物质。相应的ESI-MS谱图见图4B,仅列出四种电荷态,再次说明该物质呈天然状态寡核苷酸构象。与此HILIC谱图截然不同,图4A中相同化合物的IP-RP谱图明显“更忙碌”,共包含11种电荷态(详细信息见参考资料[3])。然而,在waters_connect软件中进行数据处理后得到的BayesSpray去卷积谱图表现出良好的质量精度(质量数误差为-4.0 ppm),如图4C中的屏幕截图所示。要达到这样的质量精度,必须在处理方法中选择硫代磷酸化(PS)寡核苷酸同位素模型,这些方法专为包含大量硫原子的寡核苷酸类型而设计。
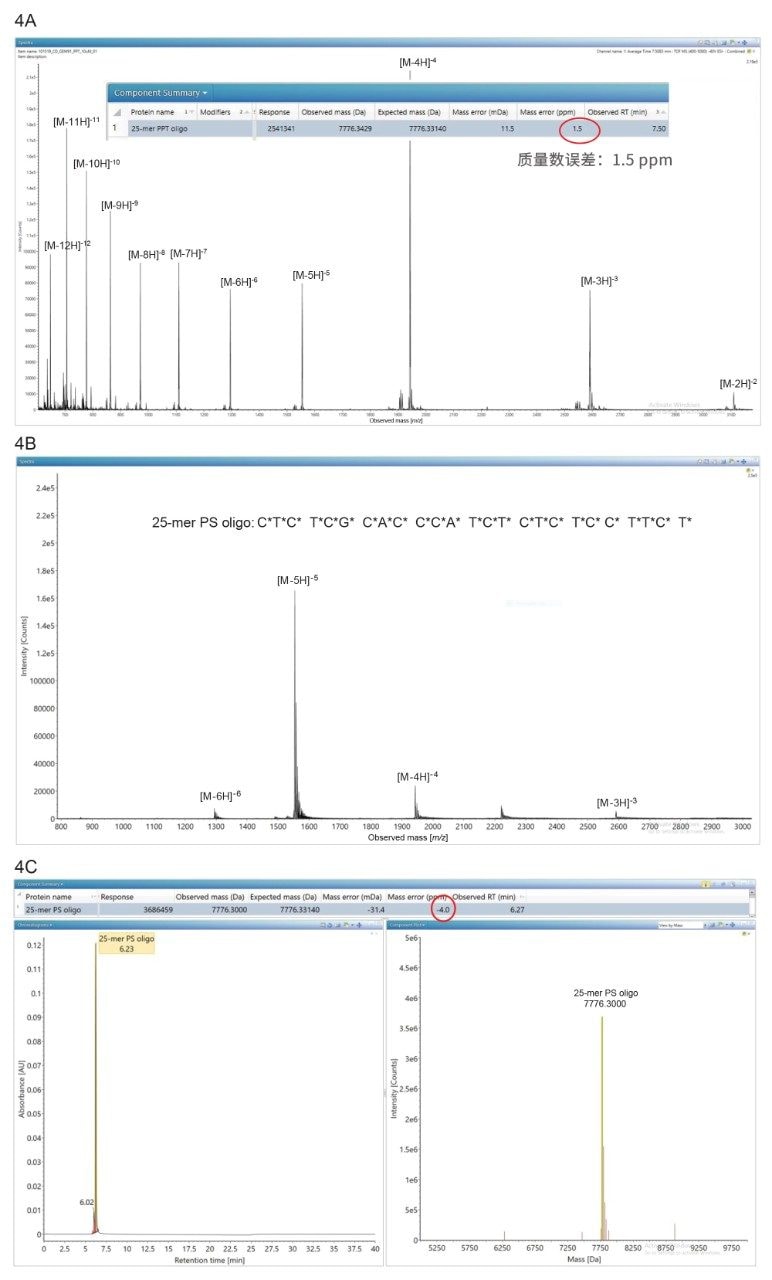 图4.(A) 25-mer PS(全硫代磷酸化)寡核苷酸的IP-RP ESI-MS谱图。插图为waters_connect处理方法中的屏幕截图,表明电荷去卷积质量数测定结果与预期质量数仅相差1.5 ppm。(B) 相同寡核苷酸的HILIC ESI-MS谱图。仅检出4种电荷态,表明该寡核苷酸呈天然状态构象。(C) HILIC ESI-MS谱图的电荷去卷积结果谱图。
图4.(A) 25-mer PS(全硫代磷酸化)寡核苷酸的IP-RP ESI-MS谱图。插图为waters_connect处理方法中的屏幕截图,表明电荷去卷积质量数测定结果与预期质量数仅相差1.5 ppm。(B) 相同寡核苷酸的HILIC ESI-MS谱图。仅检出4种电荷态,表明该寡核苷酸呈天然状态构象。(C) HILIC ESI-MS谱图的电荷去卷积结果谱图。
IP-RP与HILIC ESI-MS谱图的电荷态差异在大分子寡核苷酸中仍然可见。通过IP-RP色谱法分析57-mer寡核苷酸可得到多种电荷态(13种)(见图5A),而通过HILIC色谱法分析仅产生3种完全不同的电荷分布(见图5B)。即使HILIC ESI-MS谱图用于去卷积的电荷态较少,但在waters_connect中进行BayesSpray处理后仍可得到与IP-RP ESI-MS谱图相似的结果:质量数误差10.2 ppm vs 13.7 ppm。两种方法的完整分子量测量的质量精度均优于15 ppm。
IP-RP vs HILIC模式除了ESI-MS谱图的电荷态差异以外,新的色谱分离技术在寡核苷酸完整分子量确认的LC-MS分析成本方面也有一定优势。假设制备相同体积的流动相,IP-RP LC-MS流动相的成本通常是HILIC流动相的10倍左右。这主要是因为LC-MS级HFIP的成本高昂,这是一种具有较强毒性的改性剂,用于增加寡核苷酸的质谱响应值。HILIC流动相不需要该改性剂,因此可以避免引入IP-RP流动相相关的毒性,使该分离模式更具吸引力。此外,使用IP-RP法分析寡核苷酸时,LC-MS信号在流动相制备后24小时开始减弱,因此需要频繁制备小体积流动相(每天200~500 mL)17。 然而,UV寡核苷酸信号的稳定性不受影响,因此对于仅使用光学检测的应用而言,不需要每天制备流动相。我们通过每天使用MS和UV检测器监测dT15寡核苷酸的峰面积,研究了HILIC方法分离寡核苷酸的ESI-MS信号稳定性。我们针对HILIC流动相的洗脱液A和B制备了两份大体积(1 L)流动相,并且在2周(14天)内每天重复进样(n=3) 10 µM OST样品。我们根据OST混合物中dT15寡核苷酸三电荷单同位素质量数(m/z = 1498.57处的[M-3H]-3)的提取质谱图,通过峰面积监测ESI-MS信号。将此面积与相同寡核苷酸在260 nm处UV色谱图的峰面积比较,并将每次进样得到的MS vs UV峰面积相除,得到MS vs UV信号比。如图6中的峰面积比所示,2周内的响应值保持高度一致。此图清楚表明,HILIC流动相在本研究的时间周期内保持稳定,HILIC LC-MS分析相比IP-RP分析更具优势:HILIC LC-MS分析寡核苷酸无需频繁制备流动相。
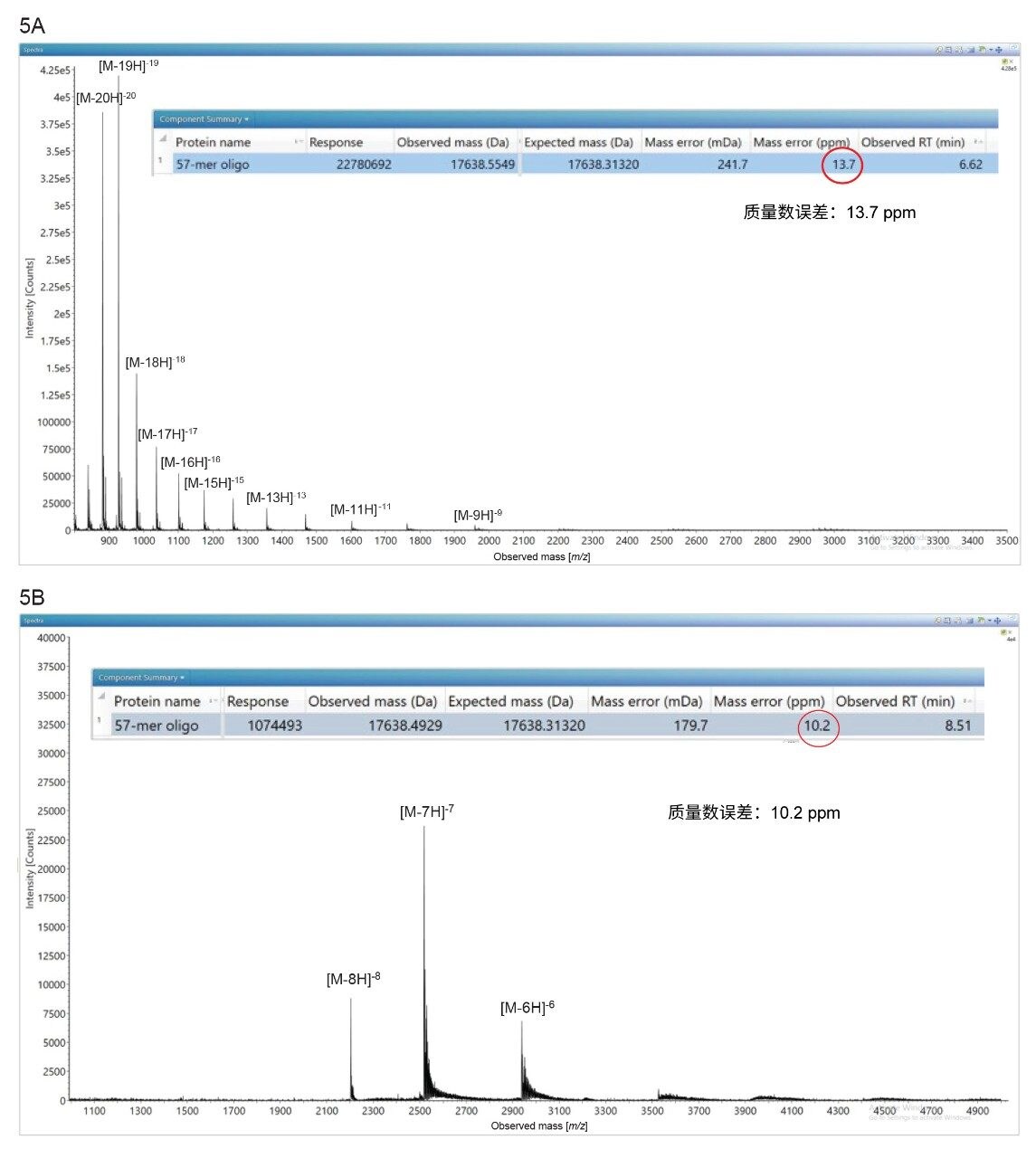 图5.57-mer寡核苷酸的ESI-MS谱图:(A) IP-RP ESI-MS谱图,显示宽分布电荷态;(B) HILIC-ESI MS谱图,仅显示IP-RP色谱法无法检出的3种独特电荷态。每种分析组合的插图突出显示了BayesSpray电荷去卷积处理后的质量数误差,两种分析方法的质量精度均低于15 ppm。
图5.57-mer寡核苷酸的ESI-MS谱图:(A) IP-RP ESI-MS谱图,显示宽分布电荷态;(B) HILIC-ESI MS谱图,仅显示IP-RP色谱法无法检出的3种独特电荷态。每种分析组合的插图突出显示了BayesSpray电荷去卷积处理后的质量数误差,两种分析方法的质量精度均低于15 ppm。
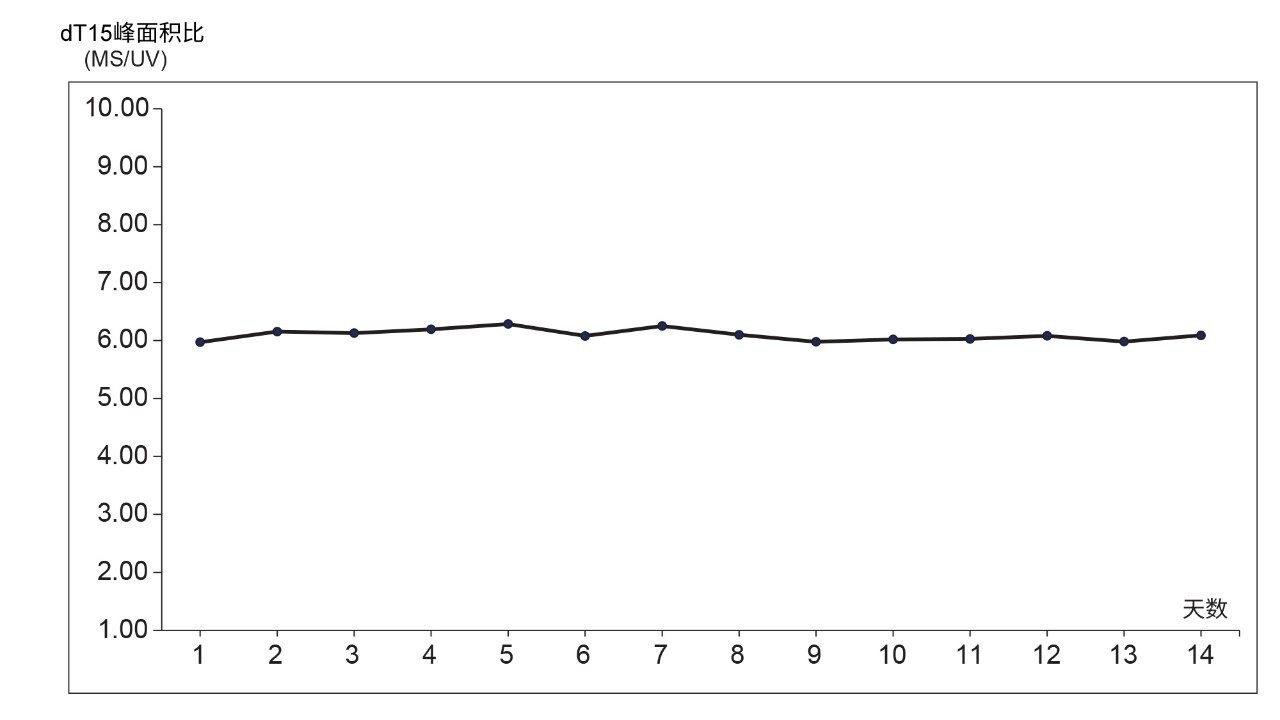 图6.HILIC流动相在2周实验期间的稳定性趋势图。此曲线反映了ESI-MS vs UV模式每天的dT15寡核苷酸响应峰面积比。对于MS响应值,使用三电荷单同位素质量数(m/z = 1498.57处的[M-3H]-3)的提取质谱图,对于UV峰面积测量值,使用260 nm处的TUV响应值。
图6.HILIC流动相在2周实验期间的稳定性趋势图。此曲线反映了ESI-MS vs UV模式每天的dT15寡核苷酸响应峰面积比。对于MS响应值,使用三电荷单同位素质量数(m/z = 1498.57处的[M-3H]-3)的提取质谱图,对于UV峰面积测量值,使用260 nm处的TUV响应值。
结论
- waters_connect工作流程经证明能够提供良好的质量精度(低于15 ppm),适合使用HILIC LC-MS分析进行寡核苷酸完整分子量确认
- 采用MaxPeak高性能表面的ACQUITY Premier BEH Amide色谱柱可以分析多达20种微量杂质,无需色谱柱钝化,且灵敏度和重现性均有提升
- HILIC LC-MS分析的优势主要集中在流动相组成方面:HILIC流动相与IP-RP流动相相比毒性低、成本低,LC-MS稳定性更佳。
- 本研究开发的HILIC LC-MS分析方法可视作寡核苷酸完整分子量分析的可行替代方法。
参考资料
- Vivek K Sharma, Jonathan K Watts.Oligonucleotide Therapeutics: Chemistry, Delivery and Clinical Progress, Future Med Chem, 2015, 7(16), 2221–2242.
- Roberts TK, Langer R, Wood MJA Advances in Oligonucleotide Drug Delivery, Nat Reviews, 2020, 19, 673–694.
- Catalin E. Doneanu, Jonathan Fox, Emma Harry, Chris Knowles, Ying Qing Yu, Joseph Fredette, Weibin Chen.利用符合法规要求的自动化LC-MS工作流程对聚核苷酸进行完整质量数确认和纯度分析, 沃特世应用纪要, 720006820ZH, 2020.
- Catalin E. Doneanu, Jonathan Fox, Emma Harry, Chris Knowles, Ying Qing Yu, Joseph Fredette, Weibin Chen.使⽤BioAccord LC-MS系统对各种经过⼤量修饰的寡核苷酸进⾏完整质量数确认分析, 沃特世应用纪要, 720007028ZH, 2020.
- Goyon A, Yehl P, Zhang K Characterization of Therapeutic Oligonucleotides by Liquid Chromatography, J Pharm Biomed Analysis, 2020, 182, 1–17.
- Sutton JM, Guimaraes GJ, Annavarapu V, van Dongen WD, Bartlett MG Current State of Oligonucleotide Characterization Using Liquid Chromatography-Mass Spectrometry: Insight into Critical Issues, JASMS, 2020, 31, 1775–1882.
- Lobue PA, Jota M, Addepalli B, Limbach PA Oligonucleotide Analysis by Hydrophilic Interaction Chromatography-Mass spectrometry in The Absence of Ion-Pair Reagents, J Chrom A, 2019, 1595, 39–48.
- M. Lauber, T. H. Walter, M. Gilar, M. DeLano, C. Boissel, K. Smith, R. Birdsall, P. Rainville, J. Belanger, K. Wyndham.Low adsorption HPLC Columns Based on MaxPeak High Performances Surfaces, Waters White Paper, 720006930EN, 2020.
- DeLano M, Walter TH, Lauber MA, Gilar M, Jung MC, Nguyen J, Boissel C, Patel AV, Bates-Harrison A, Wyndham K Using Hybrid Organic-Inorganic Surface Technology to Mitigate Analyte Interactions With Metal Surfaces in UHPLC, Anal Chem, 2021, 93, 5773–5781.
- Gilar M, DeLano M, Gritti F Mitigation of Analyte Interactions With Metal Surfaces in UHPLC, J Chrom A 2021, 1650, 462247.
- Kathryn Brennan, Mary Trudeau, Paul D. Rainville, 利⽤ACQUITY Premier系统和⾊谱柱改善
寡核苷酸⽣物分析的⾊谱性能, 沃特世应用纪要, 720007119ZH,2021. - Brooke M. Koshel, Robert E. Birdsall, Ying Qing Yu.Improving Recovery and Quantitation of Oligonucleotide Impurities Using the ACQUITY Premier with MaxPeak HPS Technology, Waters application Note, 720007238EN, 2021.
- Alpert AJ Hydrophilic-Interaction Chromatography for the Separation of Peptides, Nucleic Acids and Other Polar Compounds, J Chrom 1990, 499, 176–196.
- Hemstrom P, Irgum K, Hydrophilic Interaction Chromatography, J Sep Sci 1990, 499, 176–196.
- Gilar M, Belenky A, Smisek DL, Bourque A, Cohen AS Kinetics of Phosphorothioate Oligonucleotide Metabolism in Biological Fluids, Nucleic Acids Res, 1997, 25, 3615–3620.
- Zhang R, Diasio RB, Lu Z, Liu T, Jiang Z, Galbraith WM, Agrawal S Pharmacokinetics and Tissue Distribution in Rats of an Oligodeoxynucleotide (GEM 91) Developed as a Therapeutic Agent for Human Immunodeficiency Virus TYPE-1, Biochem Pharmacol, 1995, 49, 929–939.
- Birdsall R, Gilar M, Shion H, Yu YQ, Chen W Reduction of Metal Adducts in Oligonucleotide Mass Spectra in Ion-Pair Reversed-Phase Chromatography/Mass Spectrometry, Rapid Comm Mass Spectrom, 2016, 14, 1667–1679.
720007395ZH,2021年12月修订