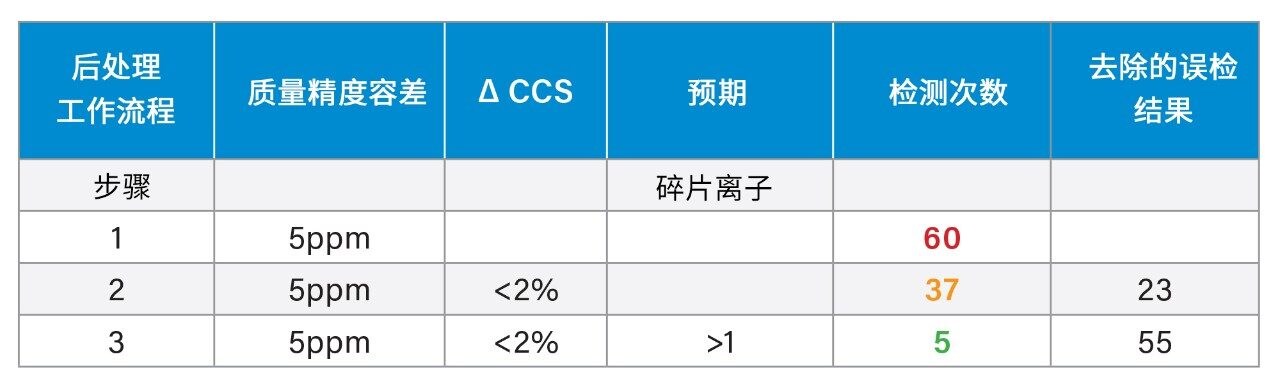
高分辨率质谱仪(HRMS),例如四极杆飞行时间质量分析器(Q-TOF),在临床、法医毒理学和代谢物鉴定中用作筛选工具越来越普遍,这些应用的目标组分都存在于复杂的生物基质(例如尿液和血液)中1,2。 使用非靶向“全扫描”数据采集,单次分析即可完成数千次检测,然后进行回顾性靶向数据分析。提高样品通量的动力非常普遍,有关提高时间效率和降低成本的要求推动了多类别化合物分析的发展。这种方法已被用于分析农药、真菌毒素、天然植物毒素3和有机污染物4,5,这些分析物也存在于各种复杂的样品基质中,从食品6到环境样品(例如污水)均涵盖在内7,8。筛查方法的目的是快速检测并鉴定所考察样品中的目标化合物,同时尽可能降低误检率。借助化合物的实测特性,例如精确质量数、同位素模式和子离子谱图,可以应用适当的筛选条件来确定样品中是否存在相应的化合物。但是,对于在复杂生物基质中仅以低浓度存在的目标化合物,单独使用这些特性来鉴定基质或分析物可能会比较困难,需要采用其他方法开发策略。对于此类复杂的分析,IM分离所提供的额外维度有助于应对此类分析挑战,并通过碰撞截面(CCS)获得更高的鉴定特异性。
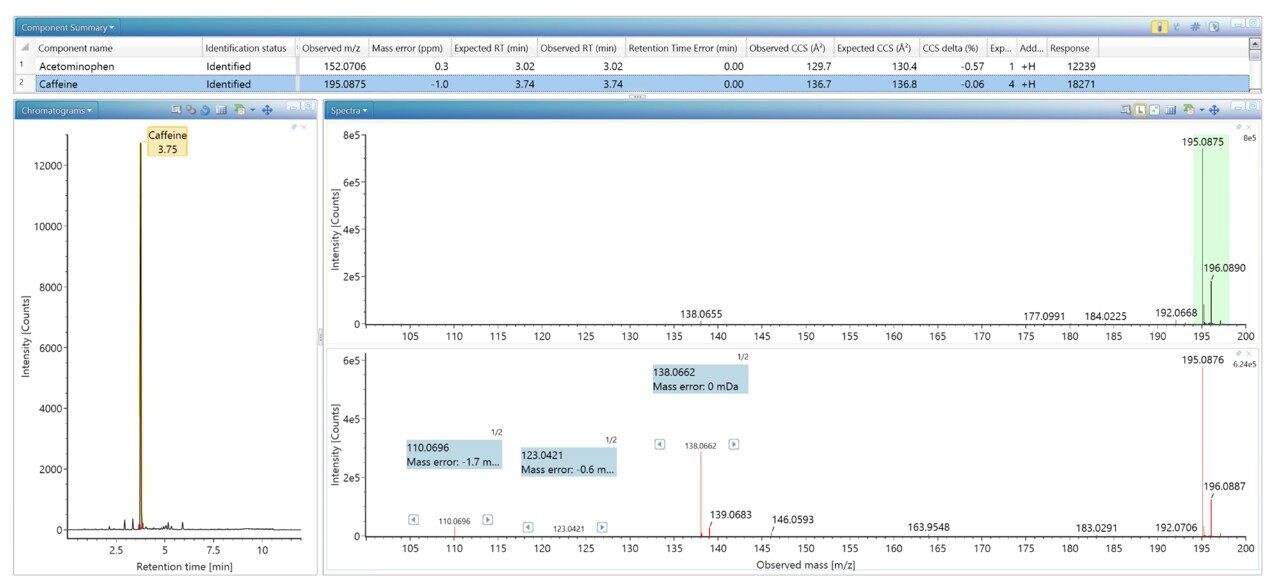
采用之前报道的质谱库生成策略9,用超高效液相色谱-离子淌度质谱(UPLC-IM-MS)表征一组获得FDA批准的市售药物。该策略能够确定保留时间(tr)、母离子、子离子和CCS。IM仪器(沃特世公司:SYNAPT (2006)、Vion (2015)、Cyclic IMS (2018))上市后,不断涌现出更多的同行评审论文(截至2014/2015年,超过1250篇)10-11,我们又开发出利用CCS作为额外终点以提高鉴定特异性的分析策略,例如农药筛查分析12。此后,CCS在小分子分析中的常规应用在多个研究领域中不断增加,包括制药(代谢、代谢组学、脂质)和食品安全(兽药、真菌毒素、类固醇、天然产物筛查、天然毒素)。生成了CCS可搜索数据库,由此可以使用CCS指标来提高鉴定的累积特异性并降低误检率。近期研究介绍了如何生成天然产物和食品添加剂数据库以及如何评价CCS测量的长期稳定性和重现性14-17。
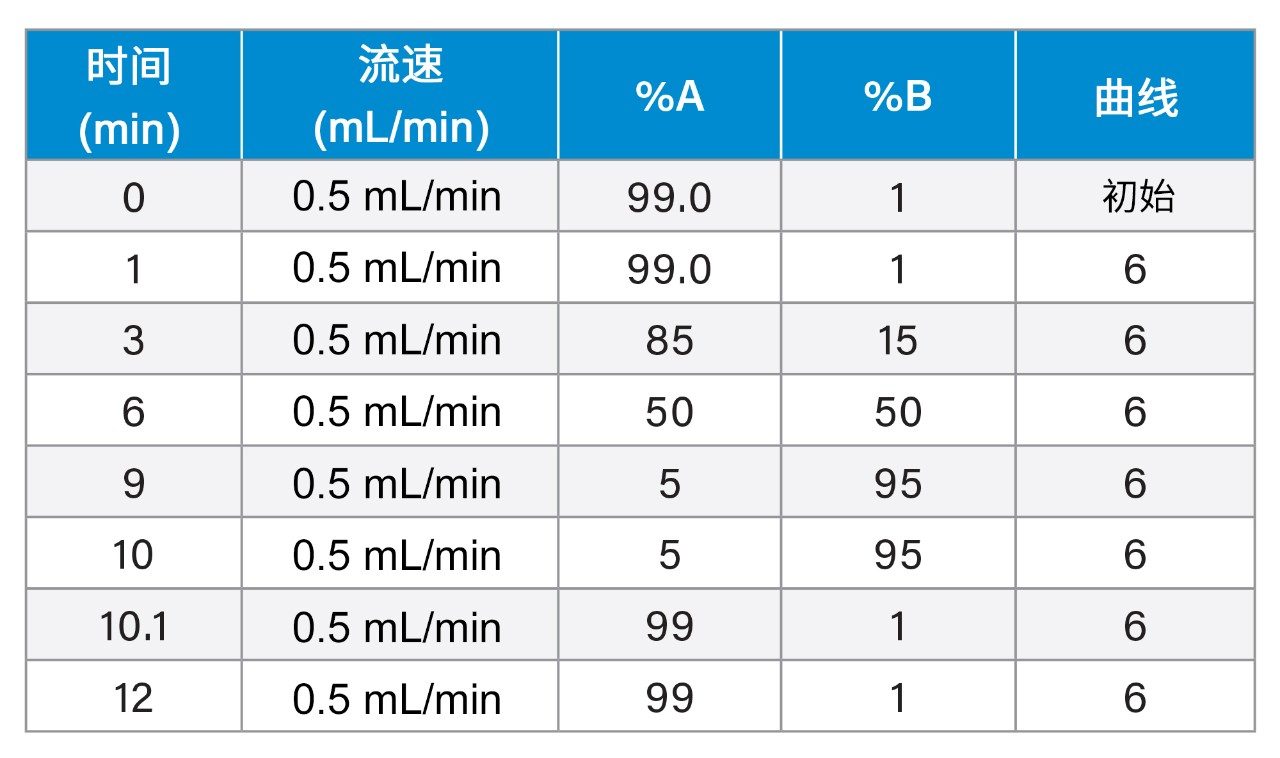
UPLC-IM包括离子淌度(MS分析之前的气相分离)和UPLC(中性物质分离)18,19。 UPLC(秒)、IMS(毫秒)和飞行时间MS(微秒)的时间尺度符合复杂样品的高通量分析要求。化合物离子淌度分离的原理是:气相离子在质谱仪中质量分析器之前充满气体的行波离子淌度(TWIM) RF离子导向装置内分离。淌度分离通过惰性缓冲气体(氮气)或使用相对较弱的电场驱动离子包来实现。离子与缓冲气体之间的碰撞次数导致漂移时间差异。由此实现的分离基于沿RF离子导向装置施加的重复DC脉冲;离子周期性地被脉冲或波超越,其中淌度较低的物质比淌度较高的物质更频繁地被超越,因此穿越装置的时间取决于淌度,并且是离子质量数、电荷和形状等因素的函数。离子淌度在LC(疏水性)和MS (m/z)之外提供了第三个分离维度。
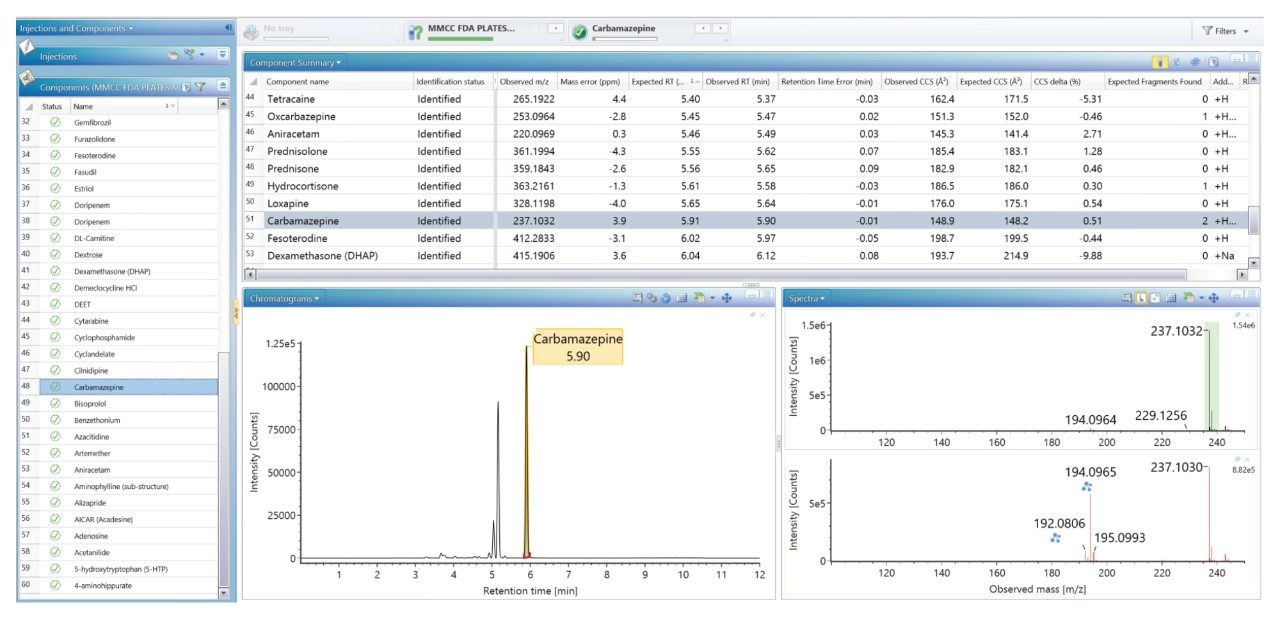
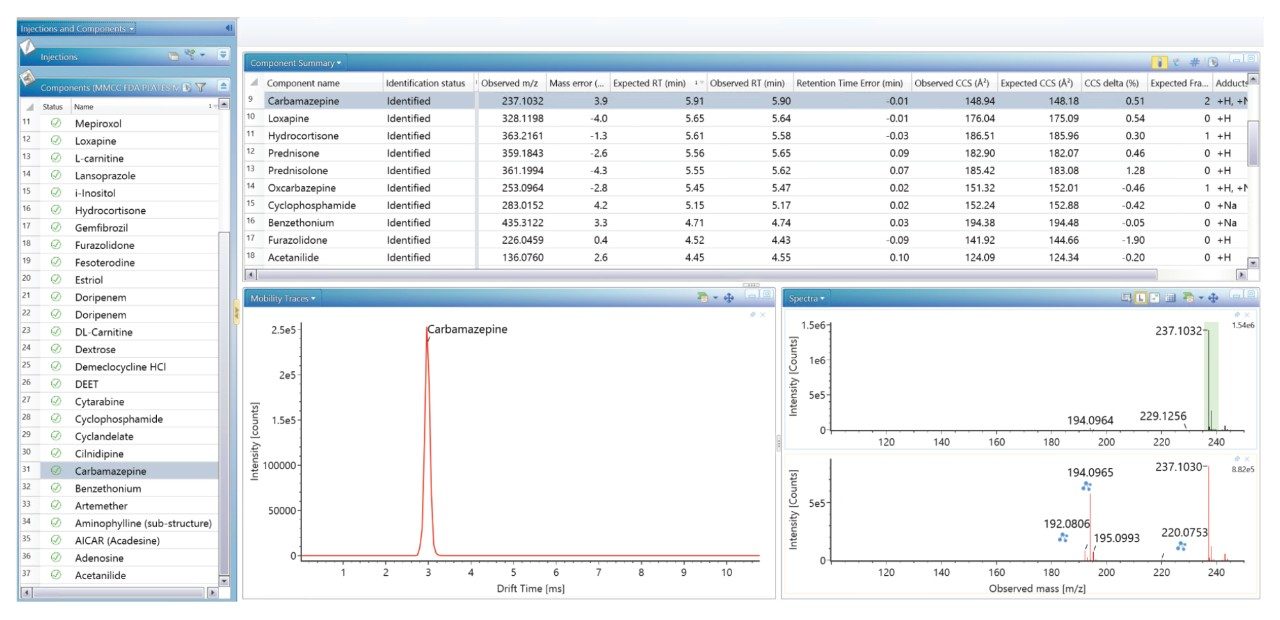
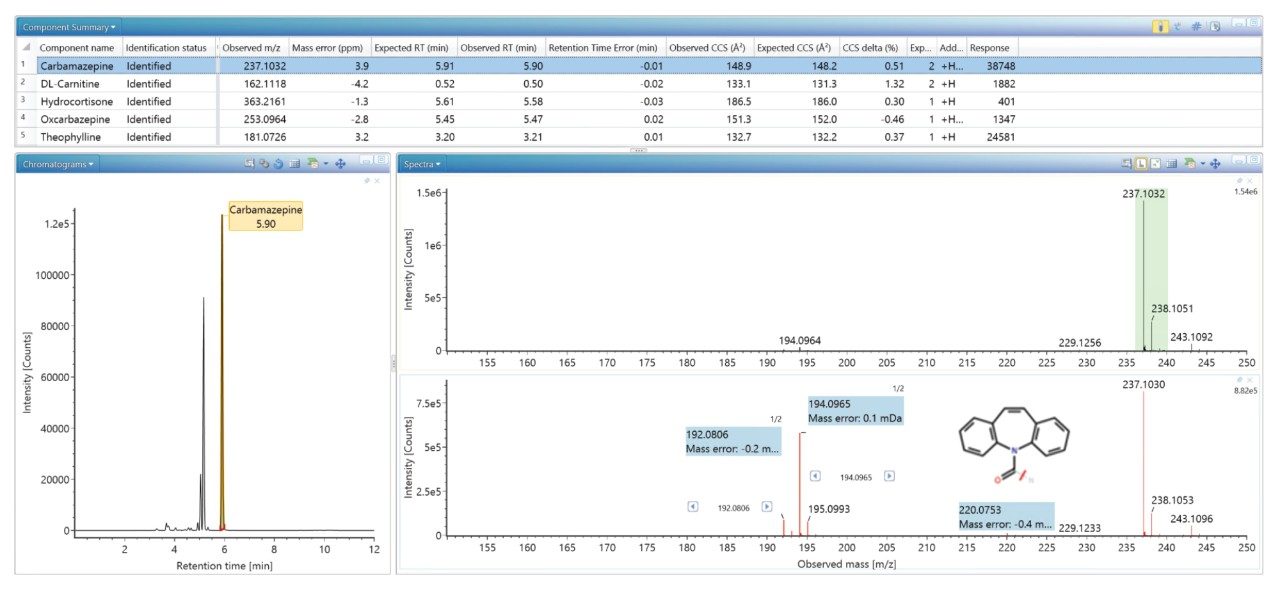
我们生成了一个包含海量FDA批准的小分子药物的数据库,并使用该数据库对患者尿样进行非靶向筛查,鉴定患者服用的药物化合物。