一种用于优化蛋白质体积排阻色谱分析的系统性快速方法开发方案
摘要
体积排阻色谱(SEC)方法的开发必须稳健有力,目标是确定可减轻不需要的次级相互作用的条件,从而根据分子体积大小产生由熵驱动的分离。蛋白质可能会发生不利的静电和疏水相互作用。采用了新型Premier SEC蛋白分析专用柱技术的色谱柱表面能够更广泛地使用这项分离技术。不过,系统性开发新的分析方法仍然值得一试。本研究提出一种23实验设计(DoE)方案,综合筛选低变量状态和高变量状态下的流动相缓冲液强度、助溶剂浓度以及柱温条件。这种立方实验设计研究所需的仪器运行时间不到2小时,并且可以使用简单的电子表格公式计算将所得结果与线性模型拟合。本研究以两种单克隆抗体和一种抗体偶联药物为例,并且在线提供对应的电子表格模板以帮助其他人加快完成他们自己的方法开发工作。
优势
- 用于优化SEC分离的快速、系统性方案
- 通过所应用的方法,只需8次实验和2小时的实验工作即可确定理想条件
- 将聚集体回收率(HMW%)、分离度(R)和峰宽(w1/2)作为析因实验设计的响应函数进行监测
简介
体积排阻色谱法(SEC)是一种用于表征治疗性蛋白质的传统技术,并被视作蛋白质聚集体常规定性和定量分析的标准方法(搭配分析型超速离心(AUC)和动态光散射(DLS)法)。该技术的优势之一是使用温和非变性的流动相条件。然而,对于这种类型的应用,SEC色谱柱内的非特异性相互作用会带来挑战。蛋白质物质和填料以及色谱柱硬件之间可能会发生不需要的相互作用,因此必须特别考虑方法参数1。 方法开发中可以使用一些通用规则,而使用析因实验设计(DoE)有助于解决非特异性相互作用的问题。
在SEC中,本体流动相和填料内部体积之间的分配效应理想情况下是由熵过程驱动的,没有任何物理化学吸附。但在实践中,这种情况少之又少。生物大分子(例如基于单克隆抗体(mAb)的药物)表现出具有不同程度疏水性和静电特性的多面性表面。因此,与填料和色谱硬件(例如色谱柱筛板)发生疏水和静电次级相互作用的可能性会对峰回收率、峰形和方法稳定性产生不利影响。为了减轻这些类型的不需要的次级相互作用,应仔细优化方法参数。
此时重点考虑两方面:(1)使用惰性色谱柱硬件和(2)优化方法参数。对于后者,可通过调整流动相中盐和有机改性剂的浓度、柱温和流速来微调方法性能2,3。 在SEC方法开发中,第一步应该是选择合适的填料。常见做法是选择平均孔径是目标分析物孔径2~3倍的填料。例如,大多数mAb的平均水合半径约为50 Å;此外,mAb聚集体的水合半径约为100 Å。因此,最适合mAb SEC分离的平均孔径通常为200~300 Å。不同物质的洗脱体积和物质之间的选择性在理想情况下仅由填料的孔径及其孔径分布确定。然而,当发生非特异性相互作用时,溶质可能会被保留并洗脱为更宽的不对称峰,从而影响分离度和聚集体定量。
要减少静电相互作用,常见方法是增加流动相的离子强度或盐浓度4,5。 也可以使用有机改性剂(例如,甲醇、乙醇、乙腈)来降低任何疏水相互作用的强度,一般通过添加5%~10%的有机改性剂来改善峰形6。 由于SEC平衡与熵有关,因此温度对平衡常数(K)和区域保留因子(k”)几乎没有影响。(请注意,传统保留因子 - k或k' - 不能应用于SEC。改用k”,它代表样品在孔内停滞的流动相或在流动的本体流动相中停留的概率之比。换言之,k”是指内部孔隙和间隙体积之间的溶质分布)。平衡常数可以根据公式1定义。
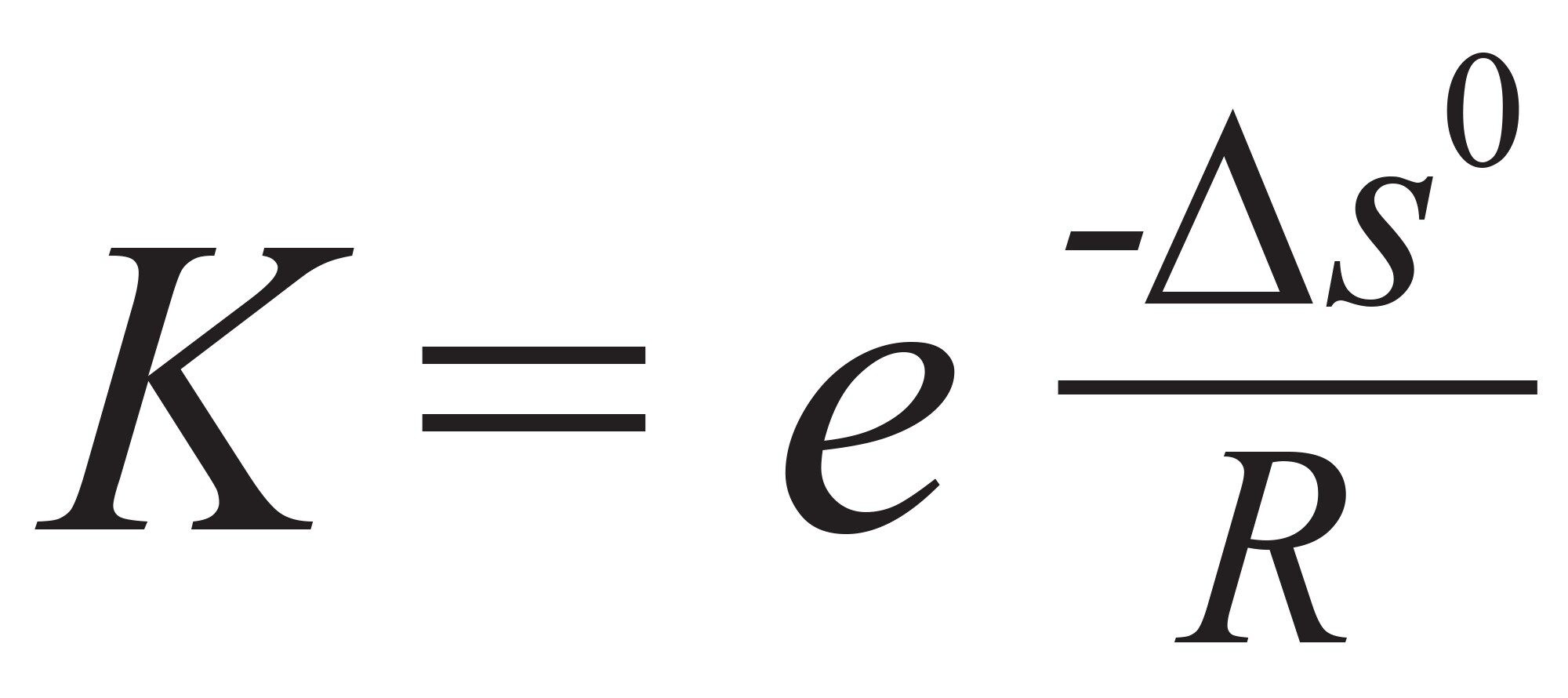 公式1
公式1
其中ΔS0是化学转变过程中的熵变,R是通用气体常数。当发生次级相互作用时,作用强度可能受温度影响,因此温度是影响(改善)回收率和/或峰拖尾的有用变量(因子)。在实践中,温度是方法优化(特别是方法控制和重现性)时需要重点考虑的一个因子。
SEC方法开发往往非常耗时,并且经常基于试错方案进行。在本应用纪要中,我们提出了一种简单的DoE来研究三个因子(盐浓度、有机改性剂浓度和流动相温度)的效应及其对SEC分离质量的影响。提供一种系统性方案快速确定理想的工作条件。
建议的因子(xj)及其水平(+/-)
1)流动相离子强度,例如磷酸盐缓冲液(PBS):1x PBS (x1(+1))和两倍稀释的(1/2x) PBS缓冲液(x1(-1)) - 根据冷泉港实验方案(Cold Spring Harbor protocol)制备7。 如今,PBS是水性SEC分离常规使用的缓冲液。不过也可以使用其他缓冲液,例如常用的磷酸钾、磷酸钠或醋酸铵溶液。使用这些缓冲液时,建议的DoE缓冲液浓度水平为50 mM (x1(-1))和150 mM (x1(+1))。
借助该因子(x1),我们可以研究静电相互作用。静电次级相互作用往往表现为在较低离子强度的流动相中HMW%降低。在较高离子强度下,HMW%增加或者单体和二聚体物质之间的选择性增加,表明静电相互作用有所缓解。
2)流动相中有机改性剂(如甲醇、乙腈、乙醇、异丙醇)的浓度,例如2% (x2(-1))和10% (x2(+1))。
借助该因子(x2),我们可以研究疏水相互作用。疏水相互作用的出现往往伴随着HMW%降低、峰拖尾或洗脱时间漂移。如果添加有机改性剂后HMW%增加、峰变尖锐或洗脱时间提前,则说明在完全水性条件下可能发生疏水次级相互作用。
3)流动相温度,例如25 °C (x3(-1))和35 °C (x3(+1)),应保持在合理范围内,以免发生变性。
借助该因子(x3),我们可以改变蛋白质的构象和扩散性以及填料表面的溶剂化。此外,可以降低非特异性相互作用的强度以提高分离质量。
也可以比较不同的色谱柱,将它们视为分类(定性)因子(xq,j)。
填料化学成分、硬件类型、平均孔径和孔径分布都会影响SEC分离的质量和HMW物质的回收率。其中每一个因子都可以被视为分类(非连续)因子。
建议监测的响应(yj)
在保留色谱模式中,通常优化的是溶质保留。市面上有多种软件包都能在成熟的半经验保留模型基础上协助进行保留建模。但是,在SEC中,溶质不会被保留,因此对保留和选择性建模不可行。与保留建模相比,应用DoE来探索最重要的因子效应可能更实用。
对于SEC – 一般来说 – 最重要的三个响应是:
1)单体与HMW之间的R (y1),
2) HMW% (y2),
3)主峰(单体)峰宽(y3)
峰对称性和回到基线的时间通常也被视为响应。然而,在大多数情况下,这些响应与HMW%回收率和峰宽密切相关,因为每一个响应都是非特异性次级相互作用的衡量指标。如果HMW%值高且/或峰尖锐,峰对称性应该也可以接受。因此,在第一步中,我们建议只研究上述响应。
建议的DoE
建议使用23全因子设计(线性模型),但也可以应用其他设计,包括部分析因设计或Plackett Burman设计,还可以应用Box-Behnken设计(非线性模型)。图1为建议的23设计示意图和对应的计划矩阵。
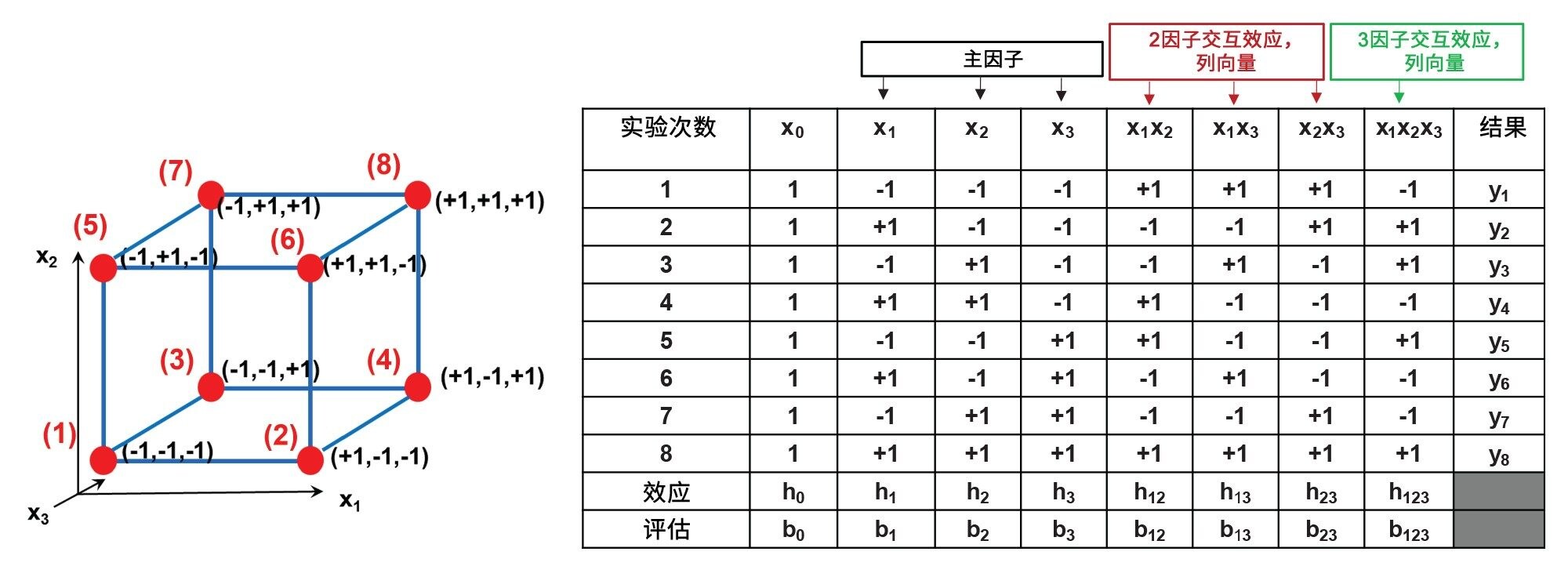 图1.23全因子设计示意图(左)和相关的计划矩阵(右)。x1–x3是所选因子,红点(1–8) – 立方体的顶点 – 显示八个实验的条件和对应的因子水平(+1或-1)。y1–y8是实验测得的响应,b0–b123是计算的模型参数。
图1.23全因子设计示意图(左)和相关的计划矩阵(右)。x1–x3是所选因子,红点(1–8) – 立方体的顶点 – 显示八个实验的条件和对应的因子水平(+1或-1)。y1–y8是实验测得的响应,b0–b123是计算的模型参数。
市面上有一些软件包可以直接计算DoE的模型参数和因子效应。在本研究中,我们鼓励用户手动计算模型和因子效应,该任务可以在Microsoft® Excel®或任何其他电子表格软件中轻松完成。
因子j的效应(h)可以计算为:公式(2)
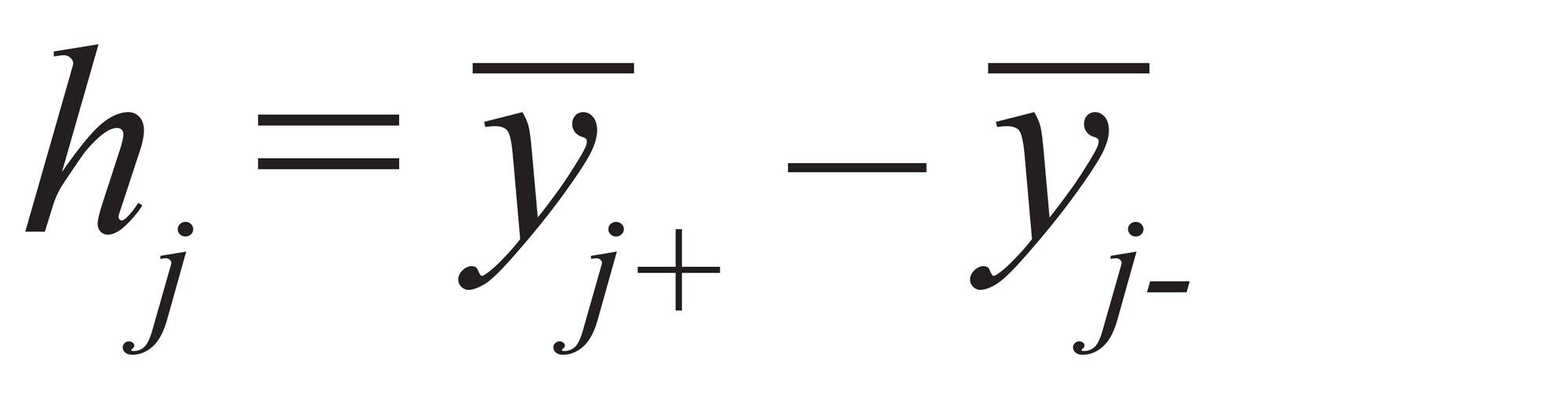 公式2
公式2
其中,yi+是在因子j的+1水平下测得的平均响应,yi-是在因子j的-1水平下测得的平均响应。
因子j的模型参数(b)为:公式(3)
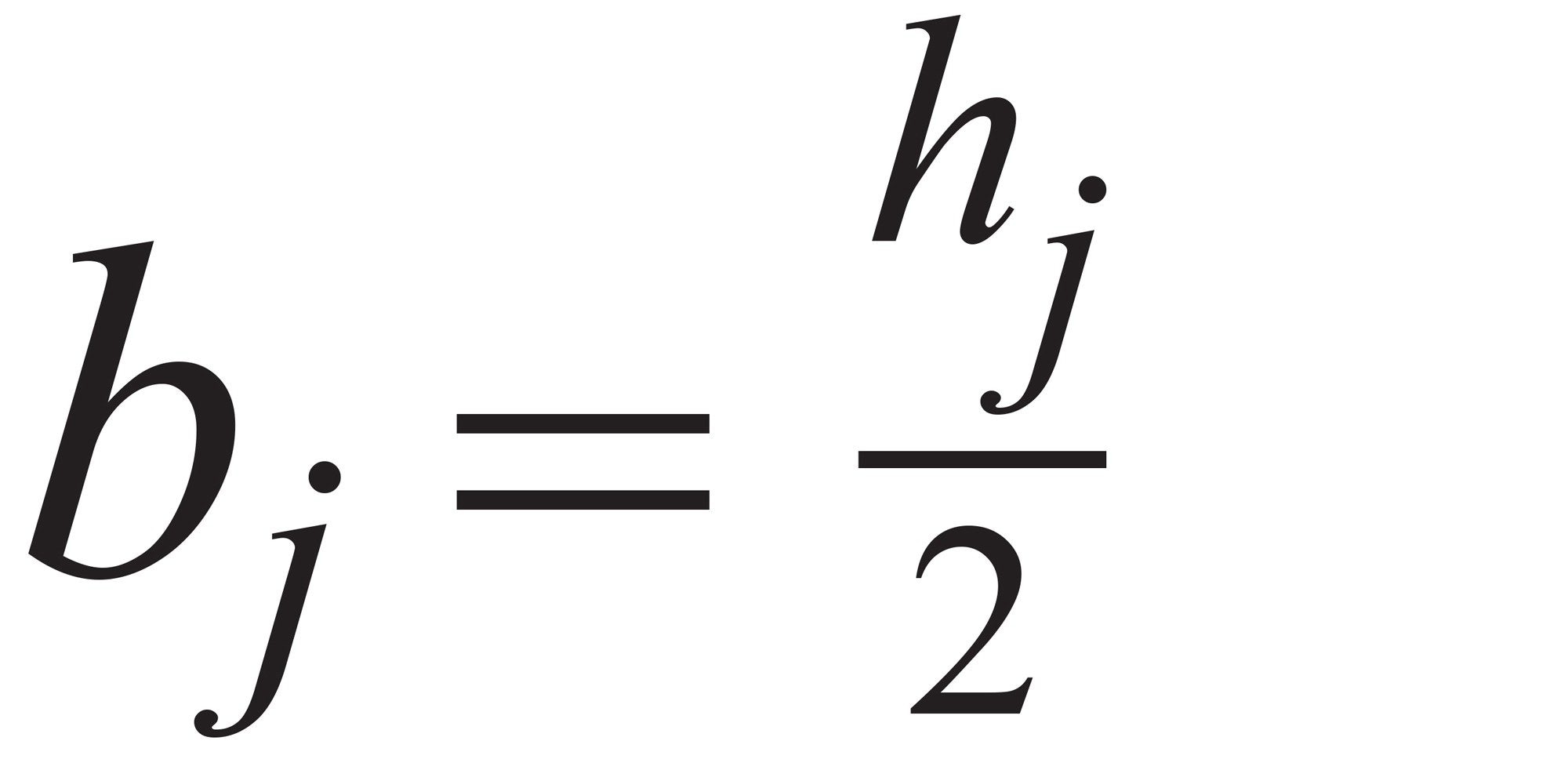 公式3
公式3
而b0参数是所有实测响应的平均值:公式(4)
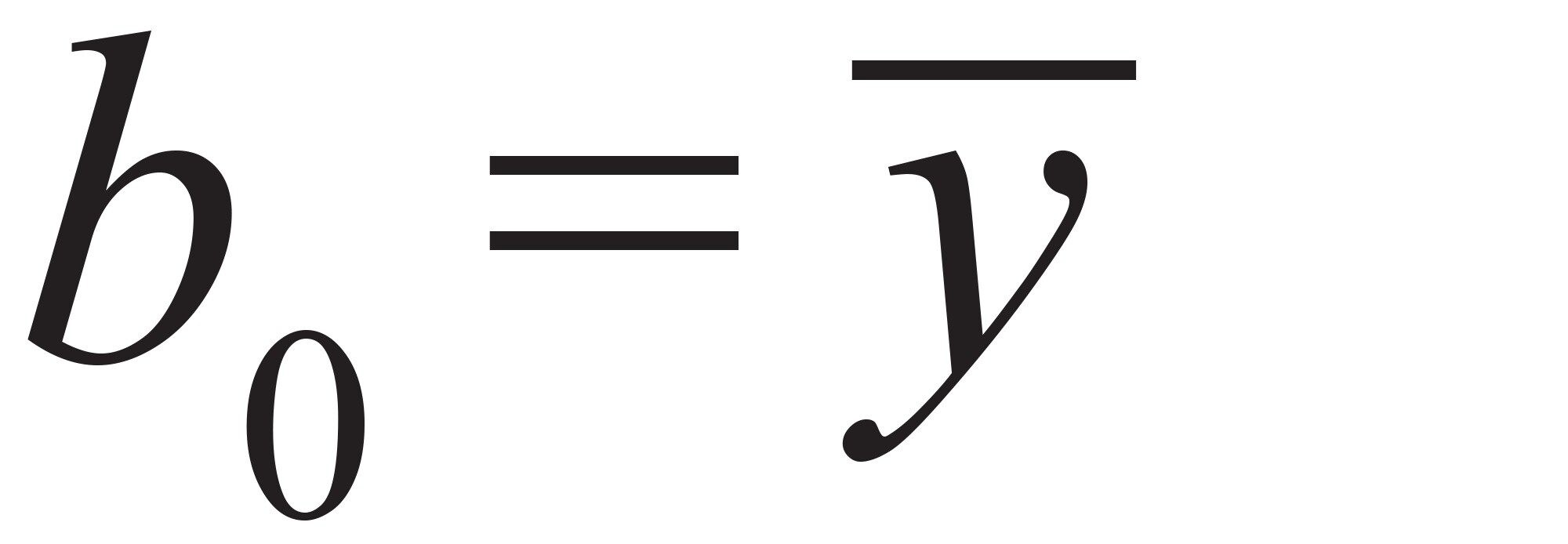 公式4
公式4
随后可编写以下模型来估算系统在任何条件下的响应:
只考虑主交互效应(一阶线性模型):公式(5)
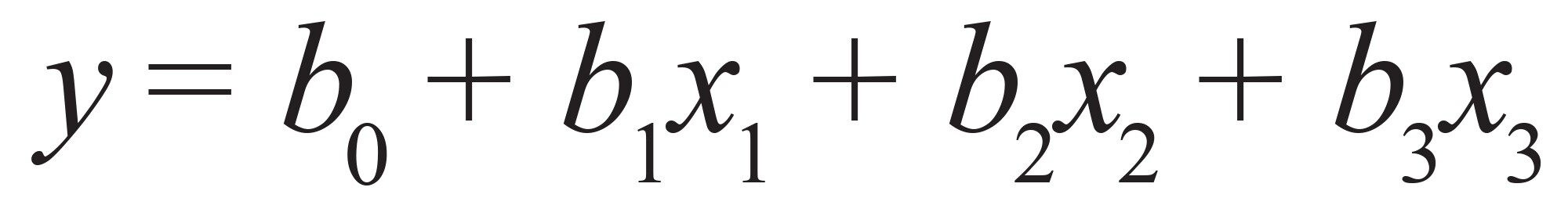 公式5
公式5
如果考虑因子交互效应(三阶线性模型):公式(6)
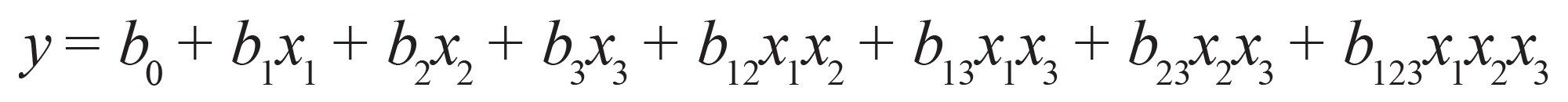 公式6
公式6
最后,因子值可以转化为因子水平。若zj为任意xj因子对应的设定值,则使用公式(7)
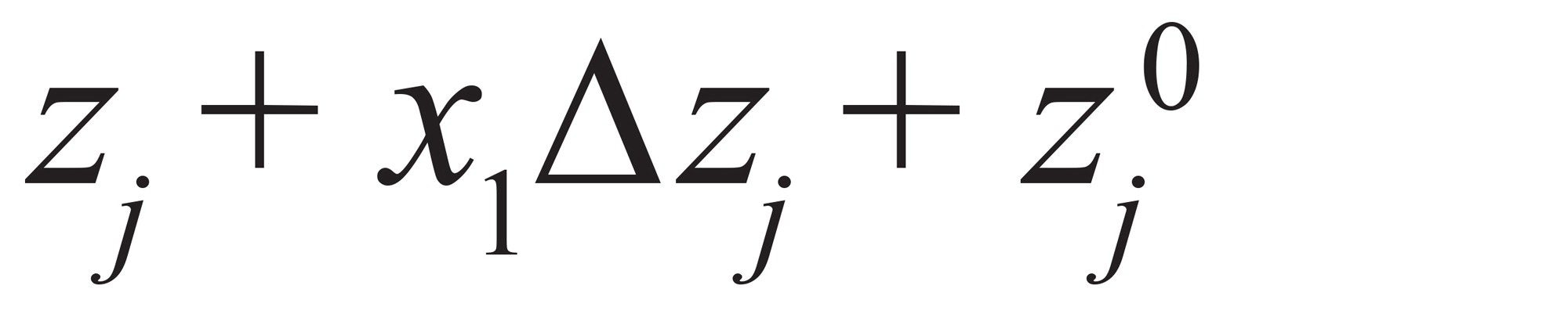 公式7
公式7
其中,Δzj是因子j在1水平的差值,zj0是因子j的平均值。
因此,因子j的水平可以计算为:公式(8)
 公式8
公式8
最后,可以利用帕累托图说明因子的效应,并使用公式5或公式6预测任何因子水平组合的系统响应。可以快速准备空白的Excel电子表格。
实验
样品和流动相制备:
利妥昔单抗(Rituxan)购自Biogen MA Inc.(美国马萨诸塞州剑桥),Kadcyla(曲妥珠单抗-美坦新偶联物)和阿瓦斯汀(贝伐单抗)购自Genentech Inc.(美国加利福尼亚州旧金山)。将样品用水稀释至1 mg/mL,然后直接进样,无需进一步前处理。HPLC级水和甲醇(MeOH)购自Fisher Scientific(爱尔兰都柏林)。磷酸盐缓冲液(PBS)各成分(氯化钾(KCl)、氯化钠(NaCl)、磷酸氢二钠(Na2HPO4)和磷酸二氢钾(KH2PO4))购自Sigma-Aldrich(瑞士布克斯)。
液相色谱条件
|
液相色谱系统: |
ACQUITY™ H-Class Bio Plus(四元) |
|
检测条件: |
UV检测(波长280 nm) |
|
样品瓶: |
聚丙烯样品瓶(P/N: 186002639) |
|
色谱柱: |
XBridge™ Premier SEC蛋白分析专用柱, 250 Å, 2.5 µm, 4.6 × 150 mm (P/N: 186009959) |
|
柱温(因子3,x3): |
25 °C(-1水平)和35 °C(+1水平) |
|
样品温度: |
8 °C |
|
进样体积: |
1.0 µL(样品) |
|
流速: |
0.3 mL/min(分析时间设置为8 min) |
|
流动相: |
pH 7.4(1x PBS和1/2x PBS),根据冷泉港实验方案[7]制备,含2%或10%甲醇。 |
|
(因子1 (x1)和因子2 (x2)): |
x1因子的水平:1/2x强度的PBS(稀释因子2;-1水平)、1x标准配方的PBS(稀释因子1;+1水平) x2因子的水平:2%甲醇(-1水平)、10%甲醇(+1水平) |
实验条件在图2中显示为23析因DoE。
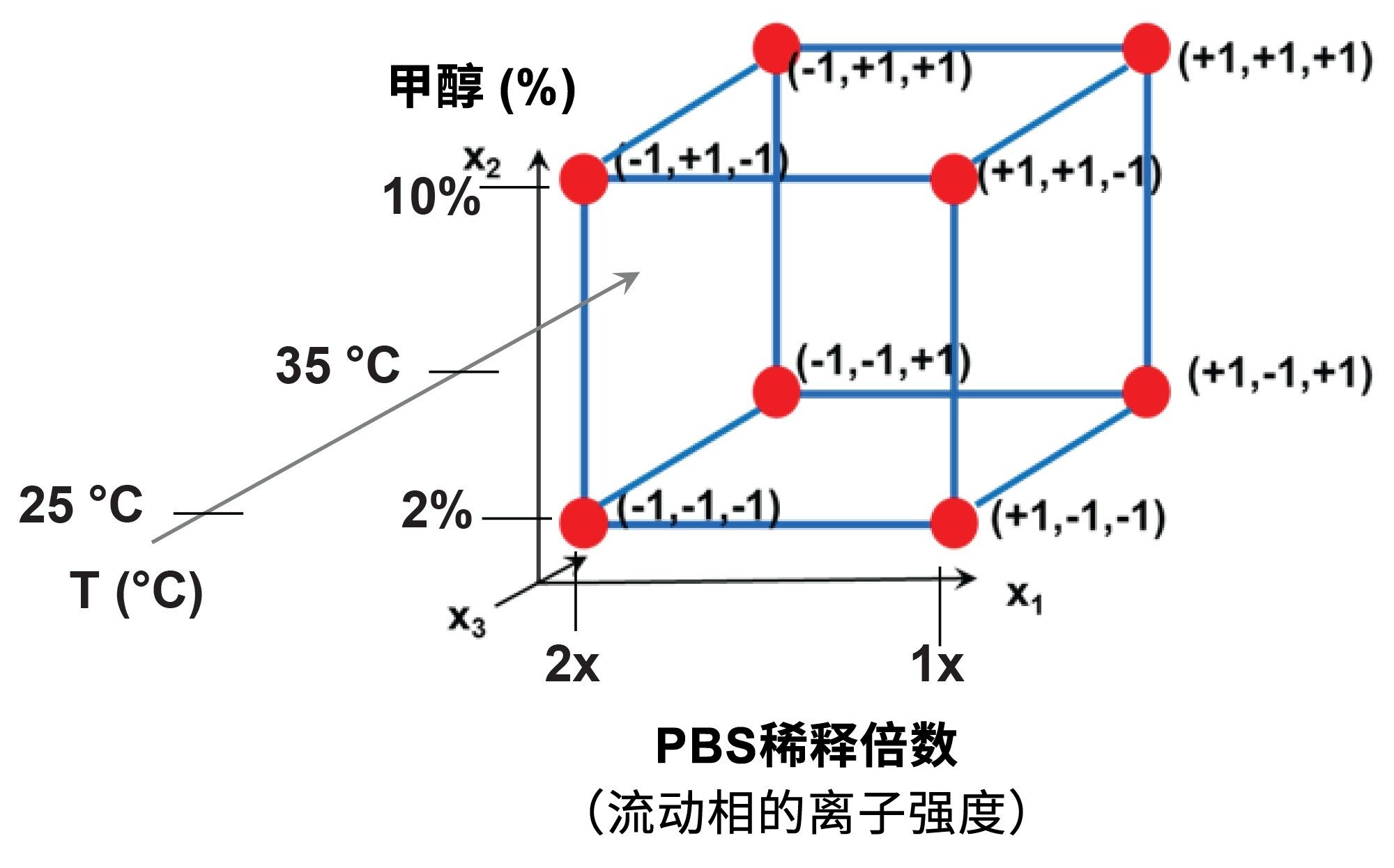 图2.DoE中设置的八个实验条件,用于估计流动相离子强度(x1因子,PBS缓冲液的稀释倍数)、流动相甲醇含量%(x2因子)和流动相温度(x3因子)的效应。
图2.DoE中设置的八个实验条件,用于估计流动相离子强度(x1因子,PBS缓冲液的稀释倍数)、流动相甲醇含量%(x2因子)和流动相温度(x3因子)的效应。
结果与讨论
本研究执行了八次实验,记录了以下响应(以利妥昔单抗为例):
- 单体与HMW之间的R (y1),
- HMW% (y2),
- 主峰(单体)峰宽(y3)
填充所有三个响应的计划矩阵,并估计因子效应。图3显示了y2响应(HMW%)的矩阵作为代表性示例。
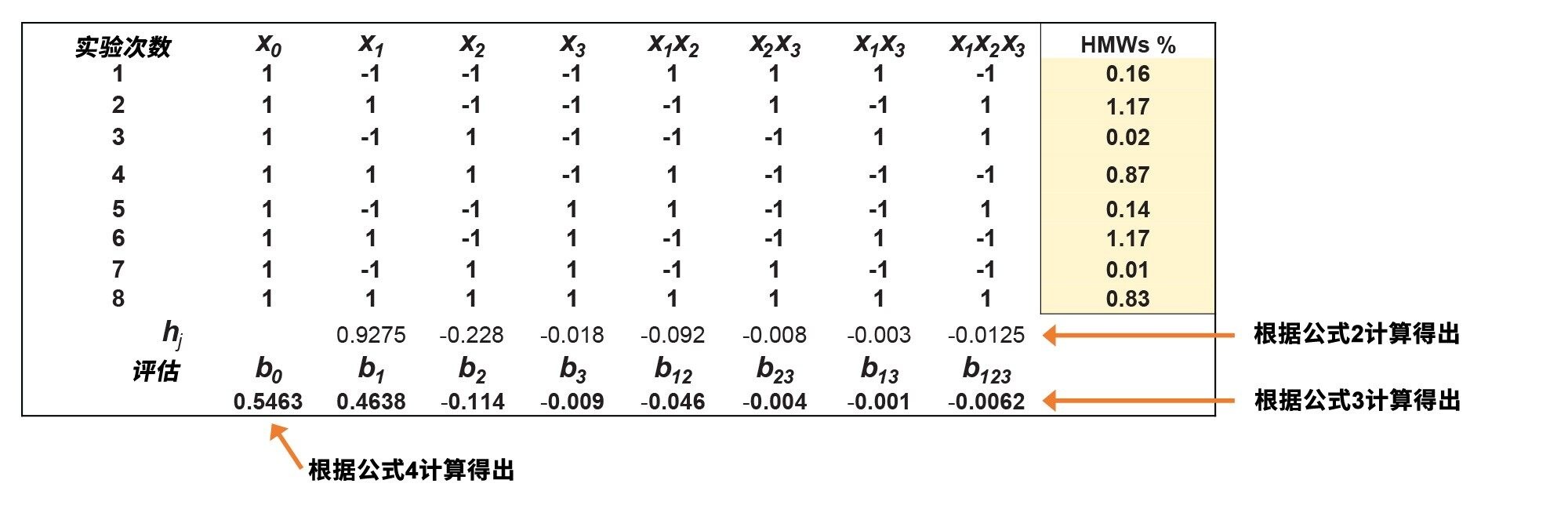 图3.y2响应的DoE矩阵(观察到的HMW%)。
图3.y2响应的DoE矩阵(观察到的HMW%)。
从矩阵中确定因子效应和模型参数。例如,对于y2因子,根据公式5和公式6得到以下模型:
如果只考虑主交互效应(公式5):公式(9)
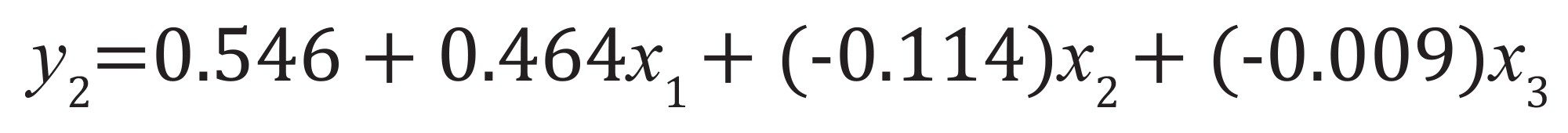 公式9
公式9
如果考虑因子交互效应(公式6):公式(10)
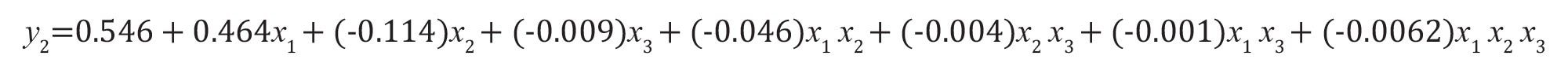 公式10
公式10
使用这些模型公式,可以估计任何方法因子组合的响应(在可接受范围内)。例如,在DoE中心点(x1=0 [z1=1.5x PBS]、x2=0 [z2=6%甲醇]和x3=0 [z3=30 °C])的y2因子(HMW%)期望值预测为y2pred = 0.546% HMW。已开展实验验证,得到y2exp = 0.550% HMW,表示与HMW%预测值有-0.7%的误差。(因子水平和值之间的转换可以通过公式7和8实现。)由于HMW%预测的偏差非常小,表明该线性模型适当。
用帕累托图来绘制因子效应非常有用。图4显示了y2响应的帕累托图。从图中可以看出,x1因子(流动相的PBS浓度)对观察到的HMW%影响最大,表明HMW物质与固定相(或色谱柱硬件)之间存在一些次级静电相互作用。流动相的甲醇含量%对HMW%影响较小,而温度和因子交互效应的影响可以忽略不计。
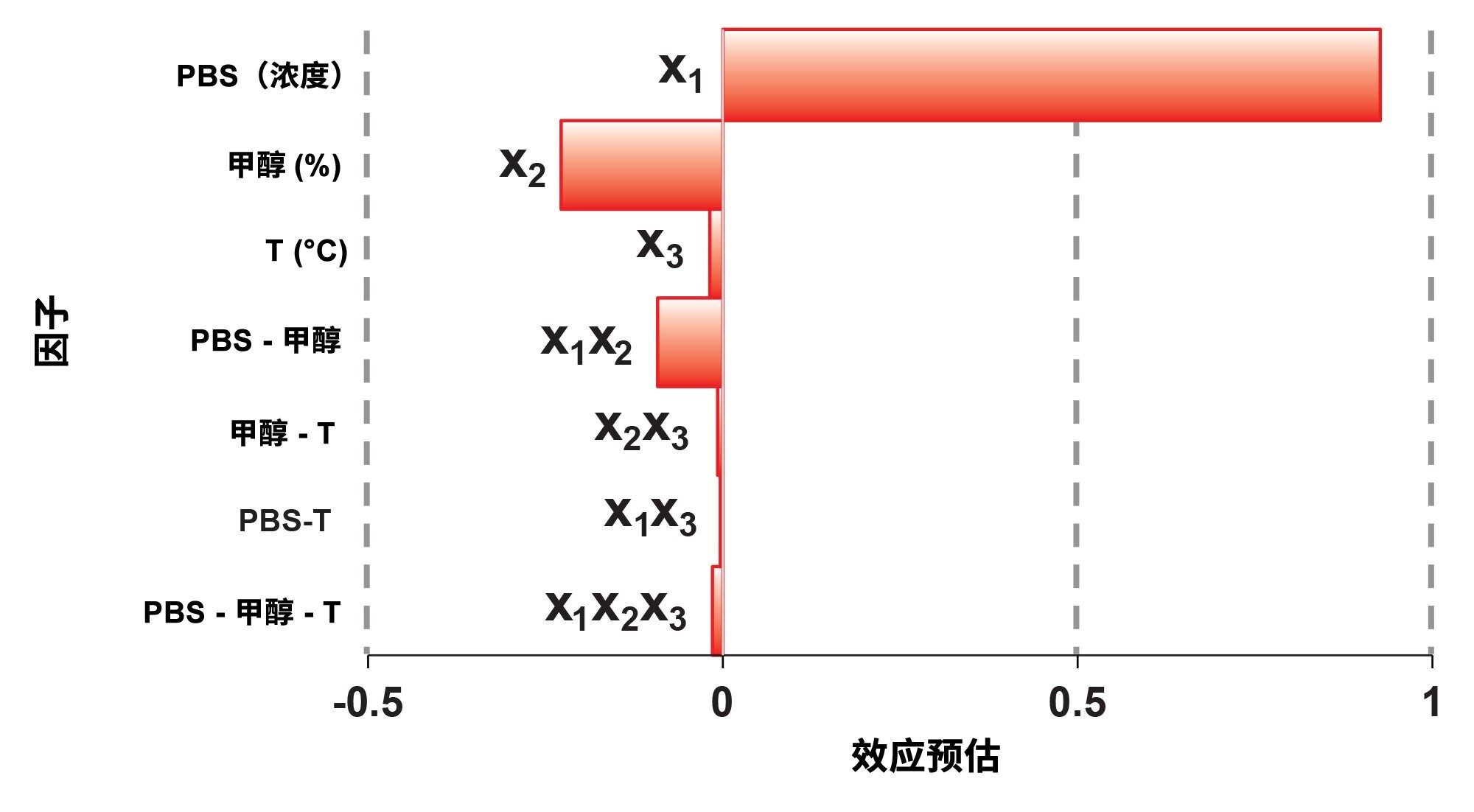
y1 (R)和y3(峰宽)响应也使用同一种方案。两个响应均受x1因子(流动相的PBS浓度)的影响最大,而y3还明显受到流动相温度的影响。最后,本研究中SEC分离的实验条件设置为1xPBS缓冲液、0% MeOH和35 °C,以获得最高的回收率和最尖锐的峰。
图5 A显示了使用与所选工作点对应的条件所获得的色谱图(理想色谱图),图5 B显示了最差条件(最低的HMW%回收率和最不对称的主峰)下的色谱图。
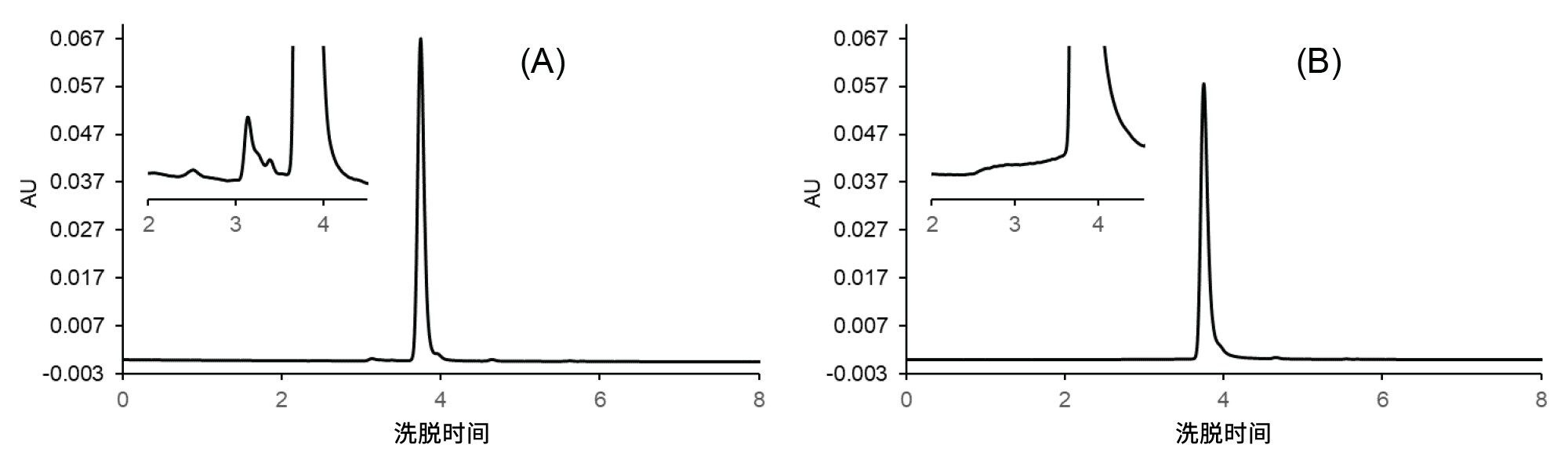 图5.实验测得的利妥昔单抗样品的SEC色谱图:使用XBridge Premier SEC 250 Å 2.5 µm 4.6 x 150 mm蛋白分析专用柱在理想条件(A)和最差条件(B)下获得的结果。
图5.实验测得的利妥昔单抗样品的SEC色谱图:使用XBridge Premier SEC 250 Å 2.5 µm 4.6 x 150 mm蛋白分析专用柱在理想条件(A)和最差条件(B)下获得的结果。
找到理想工作点后,可以调整流速以加快分析速度。在4.6 mm内径的SEC色谱柱上,选择0.3 mL/min的流速可以在分析时间和效率之间达到良好平衡,因此在最初的DoE实验中使用该流速。不过,如果目标峰之间的分离度和灵敏度足够高,可以考虑更快的流速。流速只影响SEC分离的动力学性能和光学峰检测的灵敏度。SEC的性质决定了它在较高流速下效率较低。反之,如果在最初的DoE实验中,分离度没有达到可接受的值,也可以通过降低流速来提高分离度。图6显示了使用0.7 mL/min的流速时利妥昔单抗SEC分离的加速情况。HMW物质和主峰(单体)之间的分离度仍然符合可接受标准,并且与0.3 mL/min流速下所得的结果相比,观察到的HMW%量没有变化。
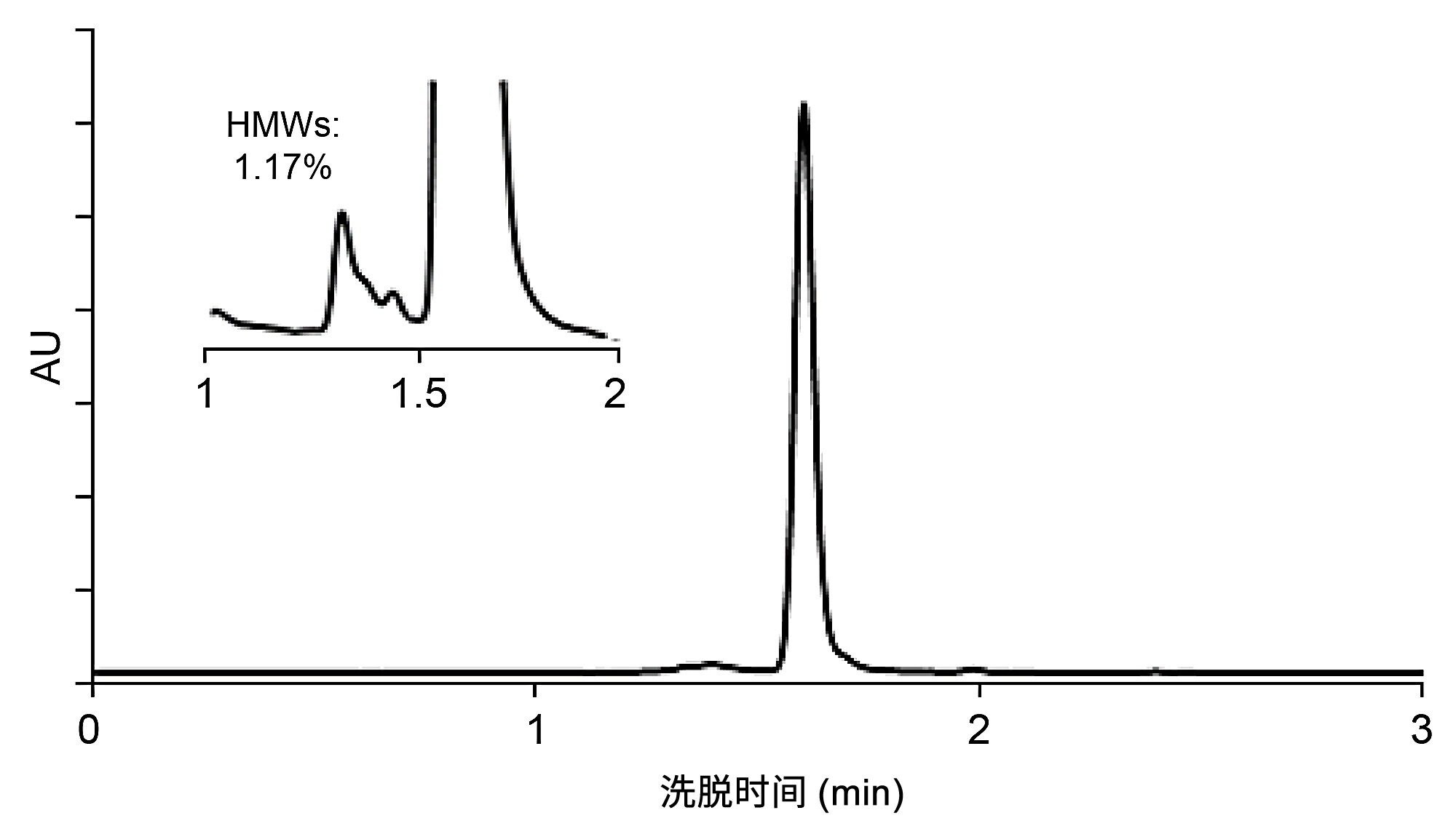 图6.在XBridge Premier SEC 250 Å 2.5 µm 4.6 x 150 mm蛋白分析专用柱上进行的快速SEC分离(利妥昔单抗)示例。流动相:1x PBS,pH 7.4,流速F = 0.7 mL/min,温度T = 35 °C,进样体积1 μL,检测:280 nm (UV)。
图6.在XBridge Premier SEC 250 Å 2.5 µm 4.6 x 150 mm蛋白分析专用柱上进行的快速SEC分离(利妥昔单抗)示例。流动相:1x PBS,pH 7.4,流速F = 0.7 mL/min,温度T = 35 °C,进样体积1 μL,检测:280 nm (UV)。
抗体偶联药物曲妥珠单抗-美坦新偶联物和mAb贝伐珠单抗也可以应用相同的DoE优化方案。找到工作点后,流速增加至0.7 mL/min。最终的色谱图如图7所示。
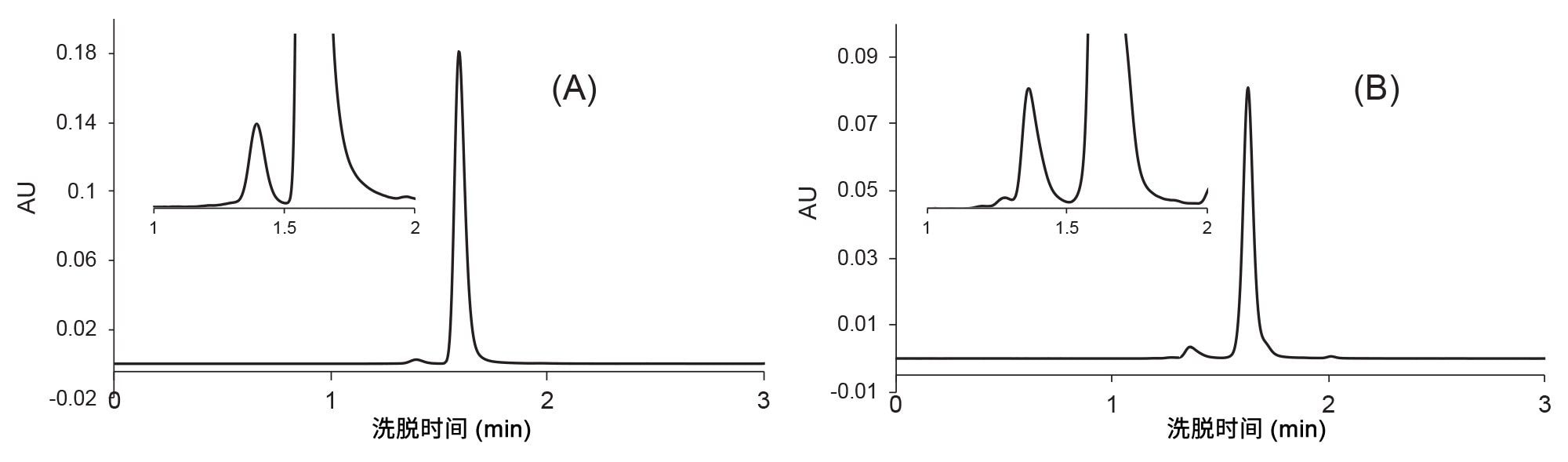 图7.在XBridge Premier SEC 250 Å 2.5 µm 4.6 x 150 mm蛋白分析专用柱上对曲妥珠单抗-美坦新偶联物(A)和贝伐珠单抗(B)进行的快速SEC分析。流动相:1x PBS,pH 7.4,流速F = 0.7 mL/min,温度T = 25 °C,进样体积1 μL,检测:280 nm (UV)。
图7.在XBridge Premier SEC 250 Å 2.5 µm 4.6 x 150 mm蛋白分析专用柱上对曲妥珠单抗-美坦新偶联物(A)和贝伐珠单抗(B)进行的快速SEC分析。流动相:1x PBS,pH 7.4,流速F = 0.7 mL/min,温度T = 25 °C,进样体积1 μL,检测:280 nm (UV)。
结论
本研究提出了一种系统性的快速方法开发方案,用于优化治疗性蛋白质的SEC分离。在SEC中没有分析物保留,因此常见的保留建模方案不适用。除常用的半经验保留模型外,我们提出了一种23全因子DoE方案来研究与分离相关程度最高的因子的效应,例如流动相离子强度(x1)、流动相有机改性剂浓度(x2)和流动相温度/柱温(x3)。仔细选择响应,并提出最有意义的响应是单体与HMW之间的R (y1)、观察到的HMW% (y2)和主峰(单体)峰宽(y3)。通过研究上述因子和响应,可以探索、理解并最终减轻静电和疏水次级相互作用。
最后,通过简单的线性模型,可以预测任何因子水平组合的响应,并确定理想工作点。
通过采用4.6 x 150 mm规格并且填充2.5 µm颗粒的低吸附色谱柱,该快速方法开发程序可以在几个小时内完成。此外,使用本应用纪要中所述的公式和公开发布的电子表格模板,用户可以轻松地重新创建和设计自己的DoE方法开发策略。电子表格模板可以从此处下载。
参考资料
- P. Hong, S. Koza, E.S.P. Bouvier, A Review, Size Exclusion Chromatography for the Analysis of Protein Biotherapeutics and Their Aggregates, J. Liq.Chromatogr. Relat.Technol.35 (2012) 2923–2950.https://doi.org/10.1080/10826076.2012.743724.
- S. Fekete, L. Kizekai, Y.T. Sarisozen, N. Lawrence, S. Shiner, M. Lauber, Investigating the Secondary Interactions of Packing Materials for Size-Exclusion Chromatography of Therapeutic Proteins, J. Chromatogr.A, 1676 (2022) 463262.https://doi.org/10.1016/j.chroma.2022.463262.
- S. Fekete, A. Beck, J.L. Veuthey, D. Guillarme, Theory and Practice of Size Exclusion Chromatography for the Analysis of Protein Aggregates, J. Pharm.Biomed.Anal. 101 (2014) 161–173.http://dx.doi.org/10.1016/j.jpba.2014.04.011.
- R.D. Ricker, L.A. Sandoval, Fast, Reproducible Size-Exclusion Chromatography of Biological Macromolecules, J. Chromatogr.A 743 (1996) 43–50.https://doi.org/10.1016/0021-9673(96)00283-X.
- M. Kamberi, P. Chung, R. DeVas, L. Li, Z. Li, X. Ma, S. Fields, C.M. Riley, Analysis of Non-covalent Aggregation of Synthetic hPTH (1–34) By Size-Exclusion Chromatography and the Importance of Suppression of Non-specific Interactions for a Precise Quantitation, J. Chromatogr. B 810 (2004) 151–155.https://doi.org/10.1016/j.jchromb.2004.07.026.
- A.M. Striegel, W.W. Yau, J.J. Kirkland, D.D. Bly, Modern Size Exclusion Liquid Chromatography, John Wiley and Sons Inc., Hoboken, New Jersey, 2009.
- Cold Spring Harbour Laboratory Press (2006), Phosphate Buffered Saline (PBS), Recipe, https://doi.org/10.1101/pdb.rec8247.
720007790ZH,2022年11月