LC-MS/MS法测定一系列植物性食品和蜂蜜中吡咯里西啶生物碱的方法开发和验证
摘要
吡咯里西啶生物碱(PA)是完全由植物生物合成的毒素。PA同时表现出遗传毒性和致癌性,且越来越多的报告显示,它在食品、草药输注液和茶中的污染相对较高。本应用纪要介绍了一种测定各种植物衍生产品(例如茶、草药、香料、孜然籽等植物产品)和蜂蜜中PA的分析方法。样品用硫酸溶液萃取,再使用Oasis MCX SPE小柱进行纯化,经过浓缩和重悬后,进行LC-MS/MS分析。本研究探讨了关键异构体对的色谱分离度。所用方法获得了良好的回收率和优异的重现性,符合CEN单一实验室验证标准中的可接受标准。单个化合物的定量限为0.6 µg/Kg,优于法规要求,因此该方法也适用于婴幼儿食品分析。
优势
- 经验证的定量方法,可分析植物性食品和蜂蜜中35种欧盟监管的吡咯里西啶生物碱,适用于法规依从性检查
- 使用Oasis MCX SPE纯化去除主要的共提物,以减少同分异构体干扰物的含量
- 使用Xevo™串联四极杆系统的特有功能RADAR,简化并加快稳定方法的开发
- 良好的方法回收率和优异的重现性,符合CEN单一实验室验证标准中的可接受标准
- 极低的定量限表明该方法有望用于分析婴幼儿产品中的吡咯里西啶生物碱,在这类产品中,PA总含量不得超过75 µg/kg
简介
PA是完全由植物生物合成的毒素,它们是抗食草动物类植物的典型次生代谢物。据估计,大约有6000种植物能够产生PA(占所有开花植物的2%)1。 截止目前,600多种不同植物中已证实存在PA,其中大部分植物来自菊科(Asteraceae/Compositae)、紫草科(Boraginaceae)和豆科(Fabaceae/Leguminosae)。近年来,越来越多的报告显示,PA污染相对较高的食品、草药输注液和茶并非由所谓的“含PA植物”制得,而主要是来自收获期间的交叉污染2。
由于PA具有遗传毒性和致癌性,被视为食品和饲料中的不良物质,因此成为2011年EFSA意见的主题3。 根据各种风险评估的结果,必须将茶/草药输注液和其他食品可能含有大量PA这一点视为相关的食品安全问题。2017年,EFSA发表了关于蜂蜜、茶、草药输注液和食品补充剂中存在PA对人类健康风险的声明4。 CONTAM专家组制定了新的行动参考值,即每kg体重每日摄入237 µg,来评估PA的致癌风险,并得出结论,认为暴露于PA可能存在相关的人体健康问题,特别是对于经常和大量摄入茶和草药输注液的消费者而言。欧盟委员会基于这些研究规定了某些食品中PA的最高含量,例如草药、香料、茶、草药输注液和花粉产品。最高含量是指35种PA的总量“下限”,相关规定见EC法规(EU) 2020/2040(自2022年7月1日起实施)5以及修订法规(EC) 1881/2006。例如,大多数茶中PA的最高含量为150 µg/kg,孜然中的最高含量为400 µg/kg。
由于PA会对人体健康构成风险,因此食品检测实验室需要开发并验证合适的确证方法来进行定量测定。目前,只有质谱检测方法为分析食品和饲料中痕量水平的PA提供了先决条件。德国联邦风险评估研究所(BfR)和负责食品和饲料中真菌毒素和植物毒素分析的欧盟参比实验室(EURL-MP)发布了用于测定植物性食品和饲料材料以及蜂蜜中某些PA组的经验证方法6-8。 但是,BfR方法不包括法规中列出的所有PA,并且存在同分异构化合物的各种共流出物;而EURL-MP方法在反相SPE纯化之前采用了中和步骤,并执行双条件色谱分析(酸性和碱性),使某些同分异构化合物获得更好的分离。当然,该分析的主要挑战是存在大量具有相同MRM通道的异构体,从色谱层面极难分离。
本研究介绍了一种使用UPLC-MS/MS定量测定植物性食品和蜂蜜中35种欧盟监管PA的简化方法,解决了在单次色谱运行中分离多种异构体的难题。本研究所提出的方法已经过内部验证,并且评估了包括正确度和精密度在内的性能参数。此外,采用RADAR证明了Oasis MCX SPE纯化方案能够增强选择性,RADAR是一种同时采集MRM和全扫描MS数据的采集模式,可在常规定量分析监测基质干扰物并且优化方法开发,是Xevo串联四极杆系统的一项实用功能。9。
实验
溶液和标准品
吡咯里西啶生物碱分析标准品购自PhytoPlan(德国海德堡)。取10 mg各化合物溶于4 mL甲醇中,制备单标储备液(2500 µg/mL)。用H2O:甲醇 95:5(v/v)制备所有35种PA的混标储备液(15 µg/mL)。使用各种样品类型的样品空白提取物连续稀释混标储备液,制备基质匹配校准物。储备液在-20 °C下储存。
样品描述和预处理
供试品包括绿茶、洋甘菊茶和博士茶的草药输注液、牛至、孜然籽和蜂蜜,均购自当地市场。为每种样品类型制备一个混合样品,用于回收率实验和基质匹配校准。萃取前,使用冲击式研磨机(IKA,英国牛津)研磨植物性商品并打成匀浆,而蜂蜜样品则在搅拌后称重并加入离心管中。
样品提取与纯化
植物性食品
茶、草药、香料和草药输注液的样品制备方法基于Kwon Y.等人(2021)10和Jeong S. H.等人(2021)的研究11。称取一部分匀浆样品(1 ± 0.01 g)加入50 mL塑料离心管中。在自动涡旋混合器上,用20 mL 50 mM硫酸的H2O:MeOH 1:1 (v/v)溶液萃取样品10 min。将样品管以5000 g离心10 min,然后取2 mL上清液加载到Oasis MCX小柱(6 cc,150 mg,部件号:186000256)上,该小柱之前已用3 mL MeOH活化并用3 mL H2O平衡。用4 mL H2O和4 mL MeOH清洗小柱。用4 mL 2.5% NH4OH的MeOH溶液将PA洗脱到硅烷化玻璃试管中(所有阶段的最大流速为1滴/秒)。然后将纯化后的提取物在50 °C温和的氮气流中蒸干,并用500 µL H2O:MeOH 95:5 (v/v)复溶。将提取物用直径13 mm的1.2 µm玻璃纤维针式过滤器过滤到TruView™ LCMS认证最大回收样品瓶(部件号:186005662CV)中以待分析。总样品稀释倍数为5倍。
蜂蜜
蜂蜜样品制备方法基于Kowalczyk E.所述的步骤提取肝脏样品中的脂质(2018)的研究12。 称取蜂蜜匀浆样品(2±0.01 g)加入50 mL塑料离心管中。用20 mL 50 mM硫酸萃取样品,振摇离心管直至样品完全溶解,随后在自动涡旋混合器上萃取10 min。将样品管以5000 g离心10 min,然后取2 mL上清液加载到Oasis MCX小柱(6 cc,150 mg,部件号:186000256)上,该小柱之前已用3 mL MeOH活化并用3 mL H2O平衡。用6 mL H2O和6 mL MeOH清洗小柱。用6 mL由乙酸乙酯:甲醇:乙腈(80:10:10, v/v/v)与1% NH4OH和1%三乙胺组成的溶剂混合物将PA洗脱到硅烷化玻璃试管中(所有阶段的最大流速为1滴/秒)。然后将纯化后的提取物在50 °C温和的氮气流中蒸干,并用1 mL H2O:MeOH 95:5 (v/v)复溶。将提取物转移至TruView LCMS认证样品瓶(部件号:186005666CV)中以待分析。总样品稀释倍数为5倍。
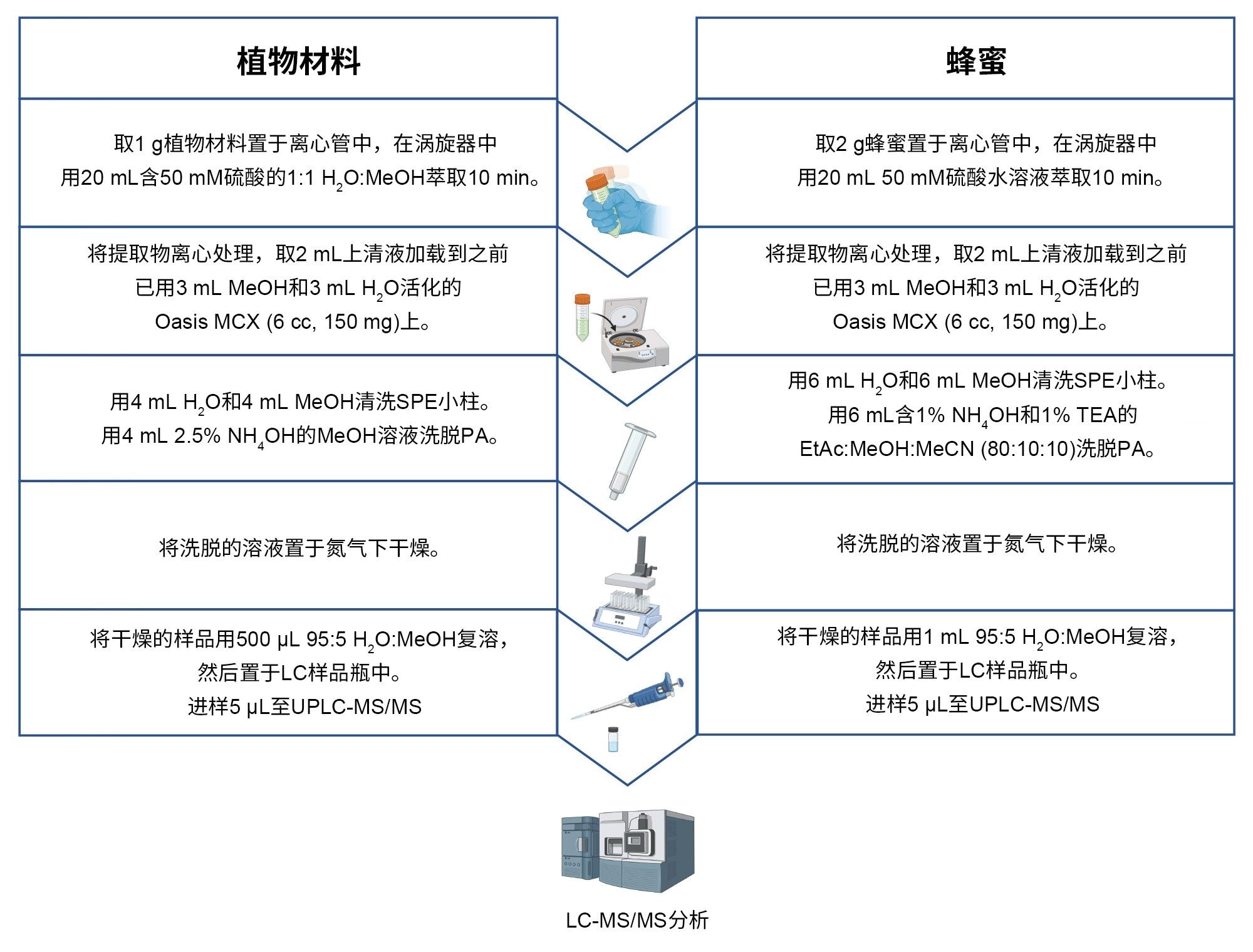 图1.样品前处理方案
图1.样品前处理方案
液相色谱条件
|
色谱系统: |
配备色谱柱管理器的ACQUITY™ UPLC I-Class PLUS系统 |
|
自动进样器: |
配备15 µL进样针的流通针式进样器(FTN) |
|
色谱柱: |
ACQUITY UPLC BEH™ C8(2.1 × 100 mm,粒径1.7 µm,孔径130 Å,部件号:186002878) |
|
水性流动相: |
含0.1% (v/v)甲酸的5 mM甲酸铵水溶液 |
|
有机流动相: |
含0.1% (v/v)甲酸的乙腈 |
|
洗针液: |
水:甲醇:乙腈:异丙醇20:40:20:20 + 0.5%甲酸(体积比) |
|
密封清洗溶剂: |
水:甲醇80:20 (v:v) |
|
柱温: |
40 °C |
|
样品温度: |
10 °C |
|
进样体积: |
5 µL |
|
流速: |
0.30 mL/min |
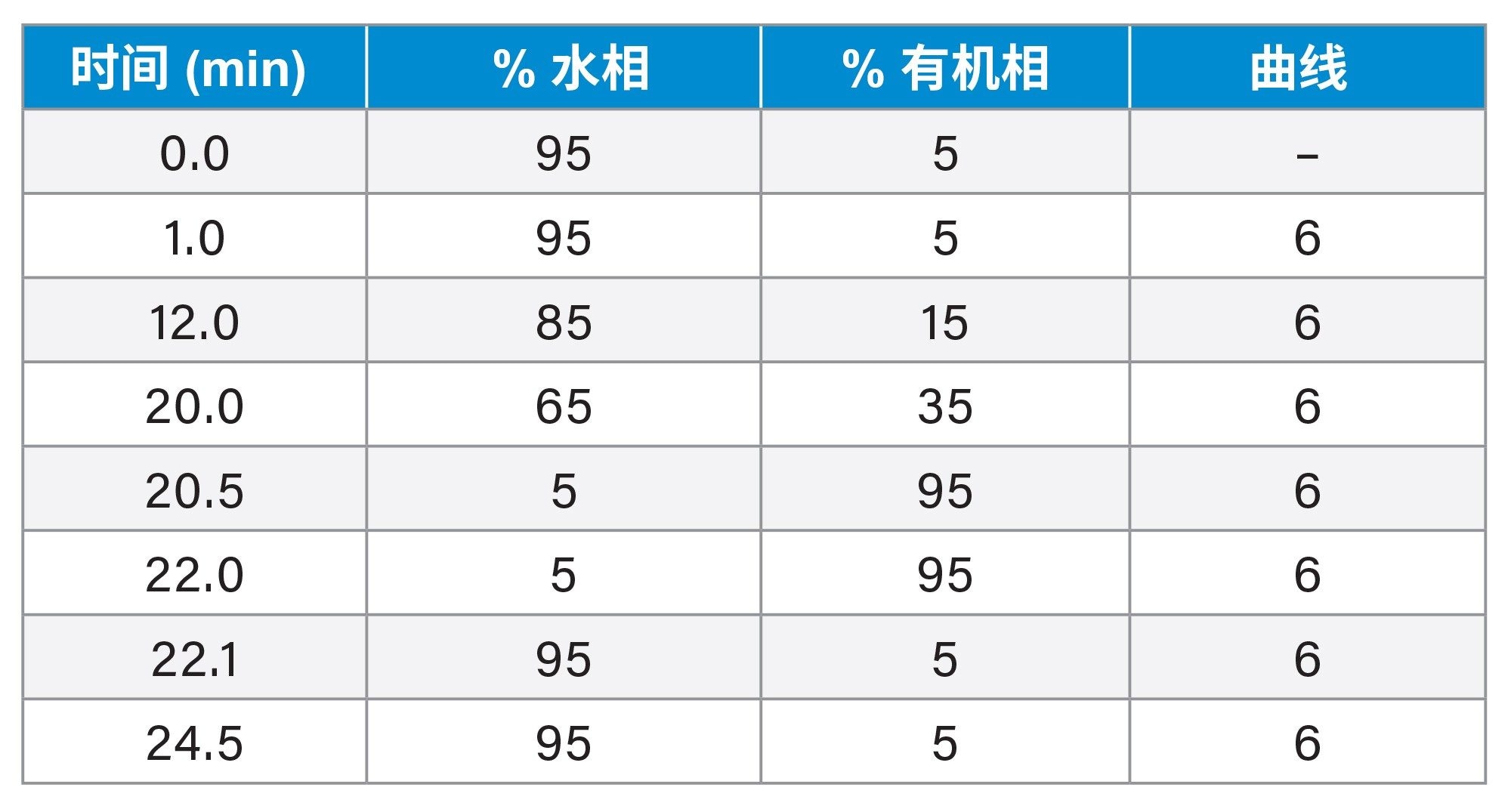 表1.UPLC梯度
表1.UPLC梯度
质谱条件
|
质谱系统: |
Xevo TQ-S micro |
|
电离模式: |
ESI+ |
|
采集模式: |
多反应监测(MRM) |
|
毛细管电压: |
+0.75 kV |
|
锥孔气流速: |
50 L/h |
|
脱溶剂气温度: |
600 °C |
|
脱溶剂气流速: |
850 L/h |
|
离子源温度: |
150 °C |
|
分辨率: |
MS1单位,MS2单位 |
|
软件: |
waters_connect™定量软件(1.0版),用于数据采集和处理 |
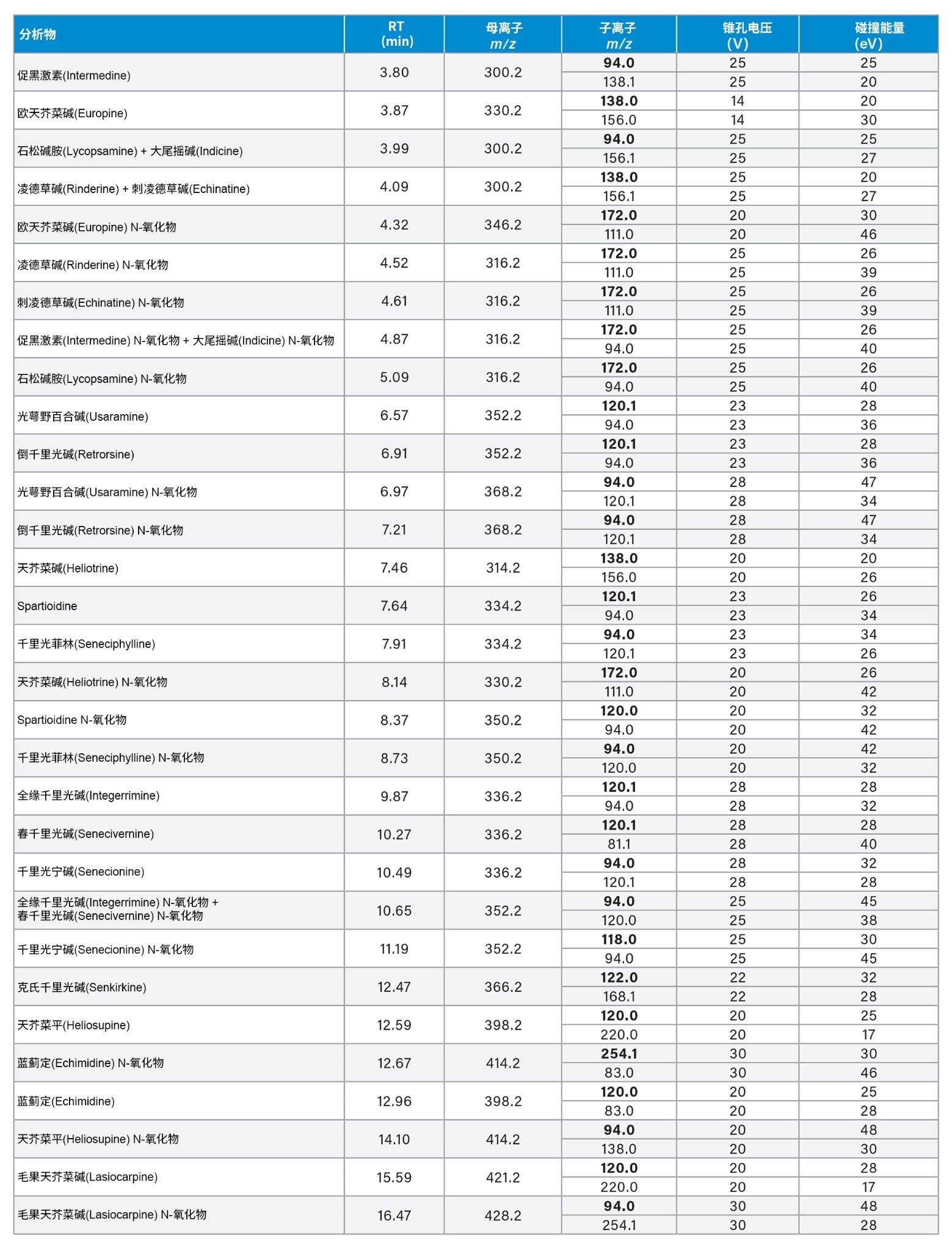 表2.PA的保留时间(RT)和优化MRM(定量离子通道以粗体显示)
表2.PA的保留时间(RT)和优化MRM(定量离子通道以粗体显示)
化合物优化
以5 µL/min的流速注入10 µg/mL各化合物溶液,同时以0.3 mL/min的流速注入流动相(1:1水相:有机相)。使用优化工具自动获取最佳母离子->子离子MRM通道、锥孔电压和碰撞能量。根据灵敏度最低的化合物对离子源电压、气体流速和温度进行优化。
优化工具可在一分钟内返回各种分析物的最佳MRM参数,分析人员只需要指定化合物名称、单同位素质量数或分子式以及待监测的加合物即可。在输出方面,该工具会生成按相对强度排序的MRM列表,分析人员可以通过交互图手动优化锥孔电压和碰撞能量(图2)。
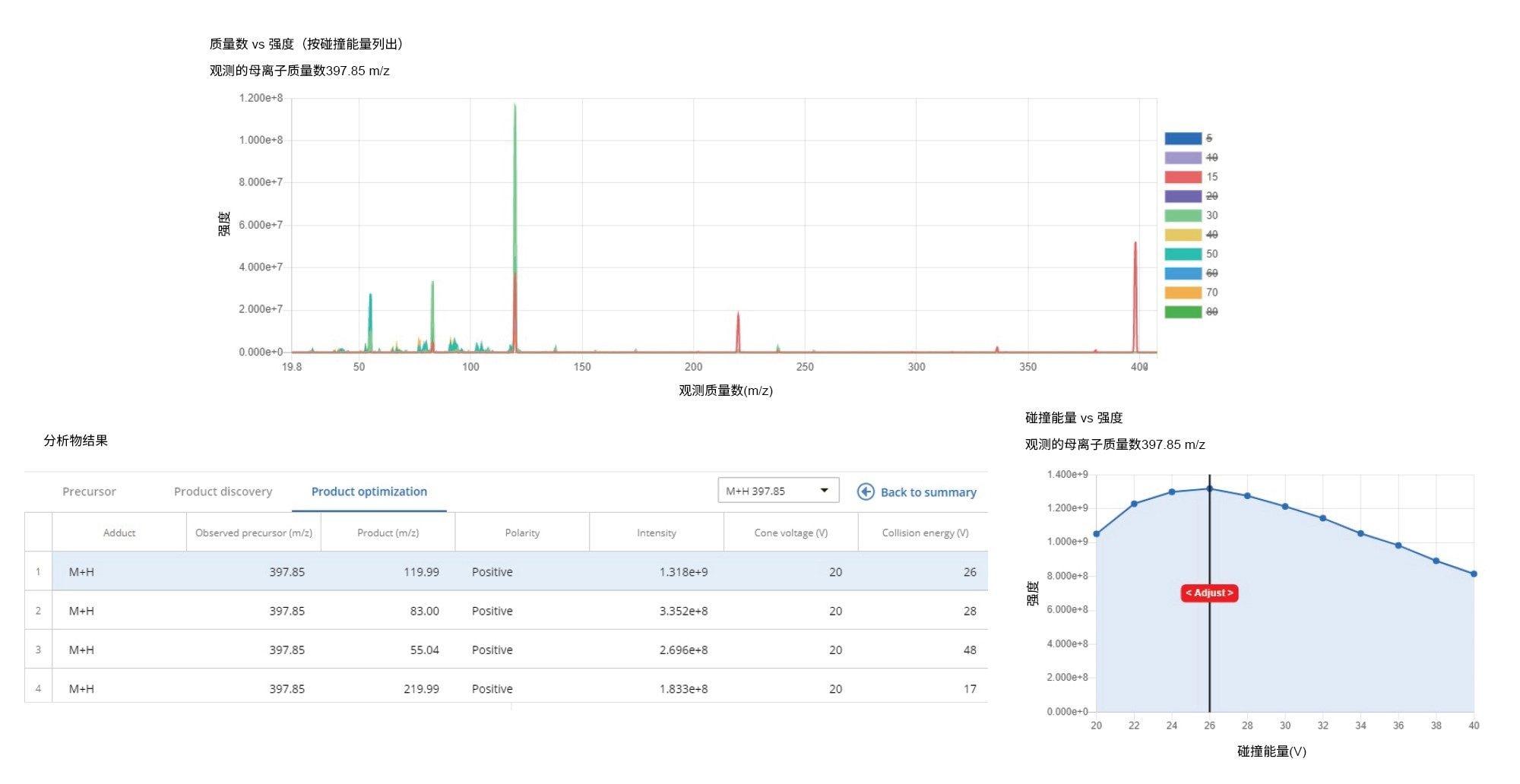 图2.优化工具输出示例:以碰撞能量为函数的蓝蓟定质谱图(上图)、前4个优化的子离子表(左下图)、子离子m/z 120的碰撞能量交互图(右下图)
图2.优化工具输出示例:以碰撞能量为函数的蓝蓟定质谱图(上图)、前4个优化的子离子表(左下图)、子离子m/z 120的碰撞能量交互图(右下图)
方法验证
通过向绿茶、洋甘菊和博士茶、牛至、孜然籽和蜂蜜的空白样品中加入35种PA的混合物评估了方法正确度,三个加标浓度为:1 µg/kg(三个重复样)、20 µg/kg(六个重复样)、250 µg/kg(三个重复样),这些浓度代表每种单独的化合物。然后按照上一节所述的方法提取并分析空白和加标样品。通过标准偏差和%RSDr评估了方法重复性。
使用文件CEN/TR 16059:2010中规定的真菌毒素方法单一实验室验证标准评价方法性能13。
结果与讨论
色谱分析
使35种PA全部实现良好的色谱分离无疑是本方法开发最大的挑战。这是因为35种化合物中共有28种存在10组异构体,还有14种化合物在反相和正相液相色谱条件下均会发生共流出现象。事实上,欧盟法规之所以对PA总量设定最大限量,而不对单个化合物设定限量,这些化合物在实现基线分离方面众所周知的难题正是原因之一。
我们在方法开发阶段测试了一系列不同的条件,旨在提高关键异构体对的分离度。
选择BEH C8 2.1 × 100 mm (1.7 µm p.s.)进行方法验证,因为它使单一方法中所有化合物的色谱分离度实现了最佳平衡。优化的液相色谱条件见实验部分。添加了35种PA的茶提取物的代表性色谱图如图3所示。以下同分异构化合物实现了基线分离:天芥菜平(Heliosupine)/蓝蓟定(Echimidine)、Spartioidine N-氧化物/千里光菲林(Seneciphylline) N-氧化物、全缘千里光碱(Integerrimine)/春千里光碱(Senecivernine)/千里光宁碱(Senecionine)、Spartioidine/千里光非林(Seneciphylline)、光萼野百合碱(Usaramine) N-氧化物/倒千里光碱(Retrorsine) N-氧化物(示例见图4)。同时我们还定量测定了四对共洗脱异构体的总量:石松碱胺(Lycopsamine) + 大尾摇碱(Indicine) (RT = 3.99 min)、凌德草碱(Rinderine) + 刺凌德草碱(Echinatine) (RT = 4.09 min)、促黑激素(Intermedine) N-氧化物 + 大尾摇碱(Indicine) N-氧化物 (RT = 4.87 min)和全缘千里光碱(Integerrimine) N-氧化物 + 春千里光碱(Senecivernine) N-氧化物 (RT = 10.65 min)。保留时间精密度优异,每个分析批次的保留时间偏移小于±0.03 min。
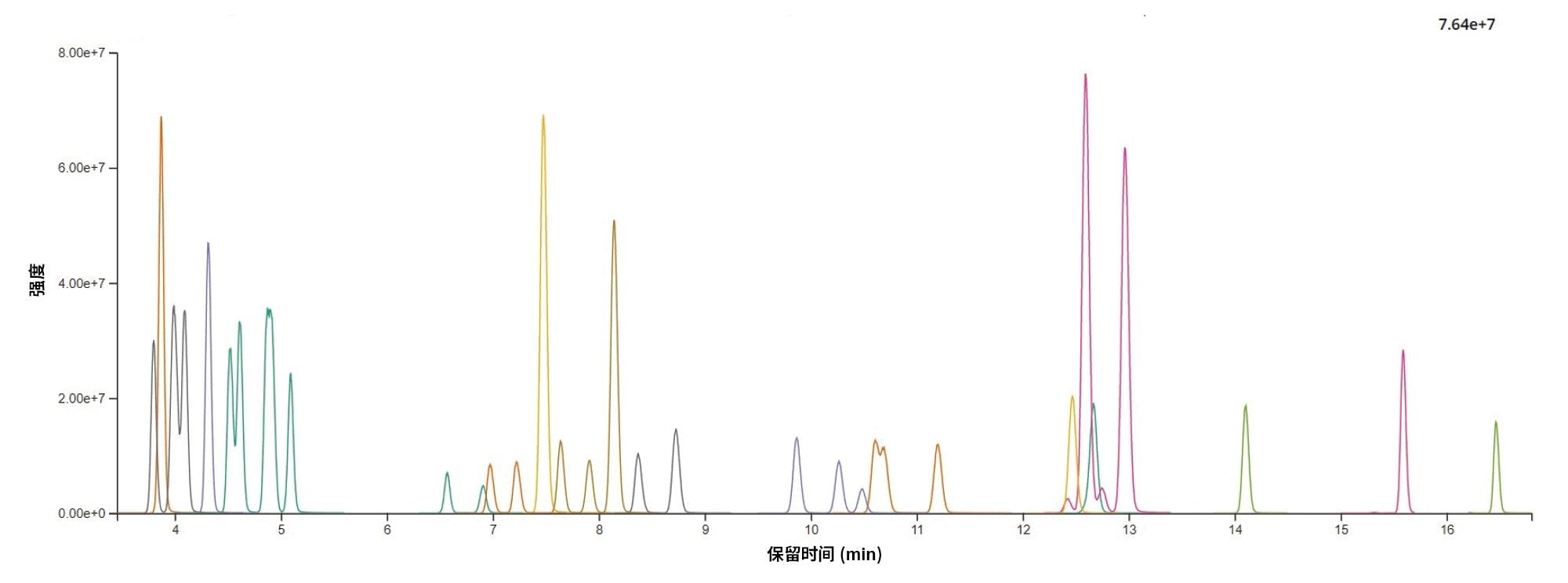 图3.35种吡咯里西啶生物碱的代表性色谱图(茶提取物的基质匹配标准品,250 µg/kg)
图3.35种吡咯里西啶生物碱的代表性色谱图(茶提取物的基质匹配标准品,250 µg/kg)
![产生以下离子的异构体的色谱峰:(a).[C18H23NO6+H]+ m/z 350.2>94.0、(b).[C20H31NO7+H]+ m/z 398.2>120.0、(c).[C18H25NO5+H]+ m/z 336.2>120.1、(d).[C18H23NO5+H]+ m/z 334.2>94.0。](/content/dam/waters/zh/app-notes/2022/720007624/720007624en-f4.jpg.82.resize/img.jpg) 图4.产生以下离子的异构体的色谱峰:(a).[C18H23NO6+H]+ m/z 350.2>94.0、(b).[C20H31NO7+H]+ m/z 398.2>120.0、(c).[C18H25NO5+H]+ m/z 336.2>120.1、(d).[C18H23NO5+H]+ m/z 334.2>94.0。
图4.产生以下离子的异构体的色谱峰:(a).[C18H23NO6+H]+ m/z 350.2>94.0、(b).[C20H31NO7+H]+ m/z 398.2>120.0、(c).[C18H25NO5+H]+ m/z 336.2>120.1、(d).[C18H23NO5+H]+ m/z 334.2>94.0。
线性和定量限
利用二次拟合和加权因子(1/x)绘制基质匹配校准曲线。对于不同类型样品的所有化合物,方法的线性范围为0.6~250 µg/kg,例外情况列于3中。对于所有化合物,校准曲线的决定系数(R2)均高于0.9900,残差均低于±20%。仪器检测限(i-LOD)14根据溶剂标准品计算得出(在LOD水平下,信噪比≥3),单个PA的i-LOD在0.001~0.02 ng/mL范围内(相当于柱上进样量5~100 fg)。如果方法的信噪比≥10,则采用方法定量限(m-LOQ)作为校准范围的最低浓度水平。共流出异构体的m-LOQ(以总和表示)为1.2 µg/kg,而单个PA的m-LOQ为0.6 µg/kg。表3显示了绿茶、洋甘菊茶、博士茶、牛至、孜然籽和蜂蜜中的i-LOD以及m-LOD、m-LOQ和线性范围。该方法的线性范围足以测定在食品监管检测中发现的典型浓度的吡咯里西啶生物碱。相比于根据同一序列校准标准品计算的平均离子丰度比,所有被测样品的离子丰度比偏差均在±30%以内。
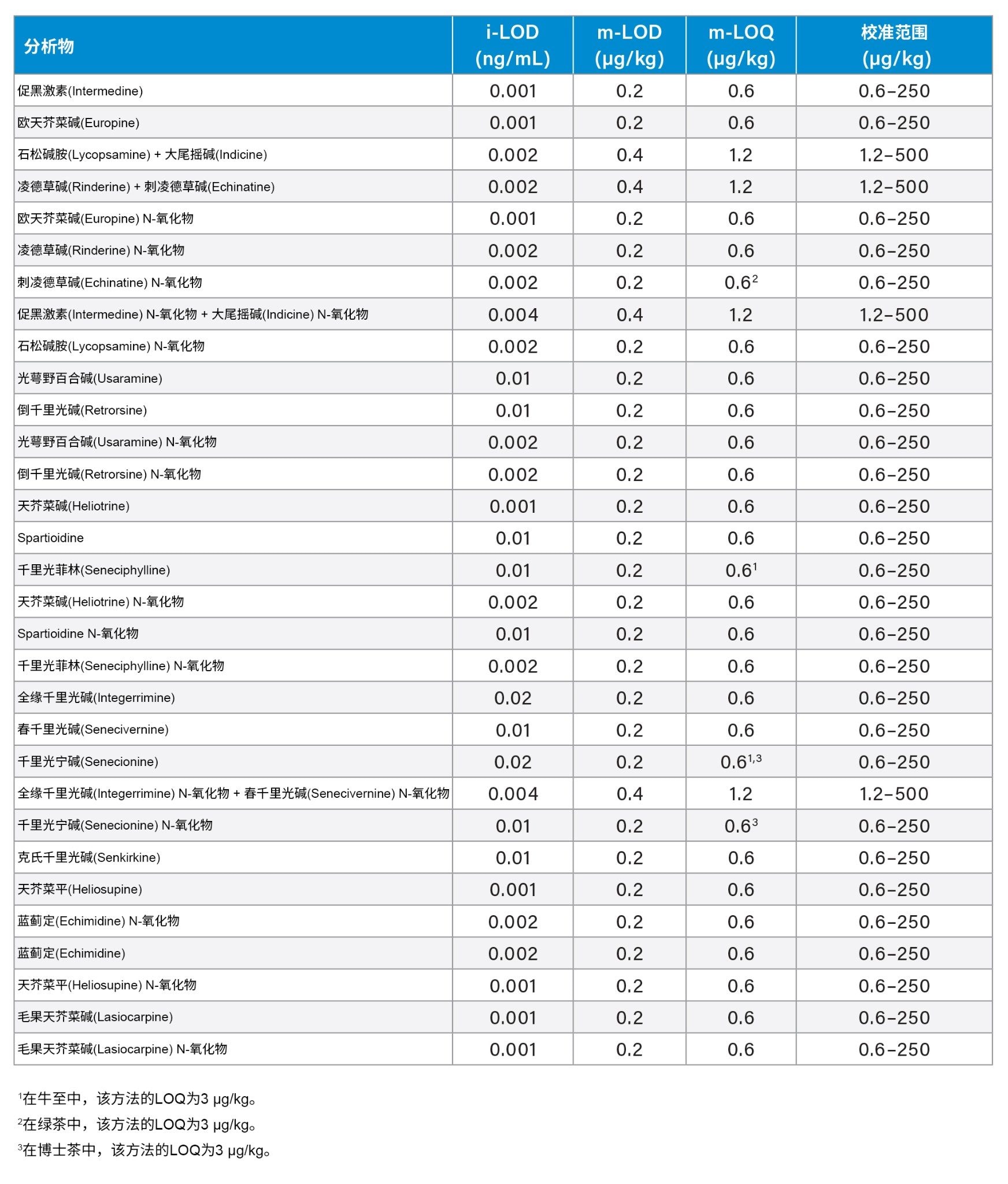 表3.Xevo TQ-S micro的仪器检测限(i-LOD),PA分析方法检测限(m-LOD)、定量限(m-LOQ)以及线性工作范围
表3.Xevo TQ-S micro的仪器检测限(i-LOD),PA分析方法检测限(m-LOD)、定量限(m-LOQ)以及线性工作范围
正确度和重复性
通过向空白样品中添加三个浓度标准品的回收率实验评估了正确度。综合考虑整个PA化合物组时,所有加标浓度下的平均回收率分别为:绿茶85% ± 10%,洋甘菊茶91% ± 8%;博士茶92% ± 10%,牛至87% ± 10%,孜然籽103% ± 6%,蜂蜜77% ± 10%。植物性样品中所有PA的回收率均在62%以上,而蜂蜜中刺凌德草碱(Echinatine) N-氧化物、欧天芥菜碱(europine) N-氧化物、石松碱胺(Lycopsamine) N-氧化物和凌德草碱(Rinderine) N-氧化物的回收率在50%~60%之间。但是,由于所有加标浓度下所有化合物在重复性评估条件下的RSD%均低于10%,因此可以应用回收率校正因子,精密度不会受影响。或者,就蜂蜜而言,可以使用程序校准来补偿回收率损失。结果重复性良好,在所有情况下RSDr%大多低于10%,不超过20%。回收率如表4和图5所示。
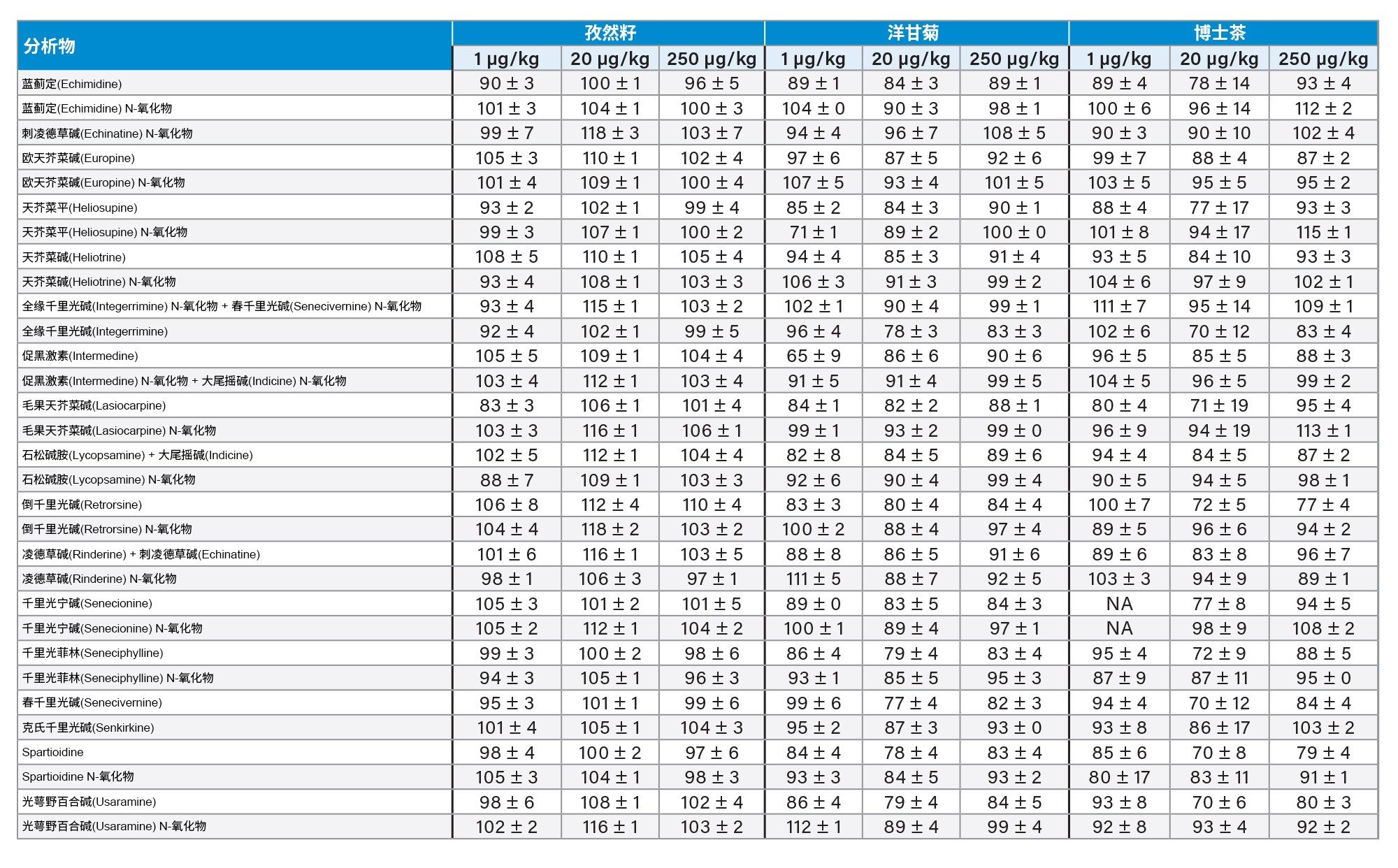
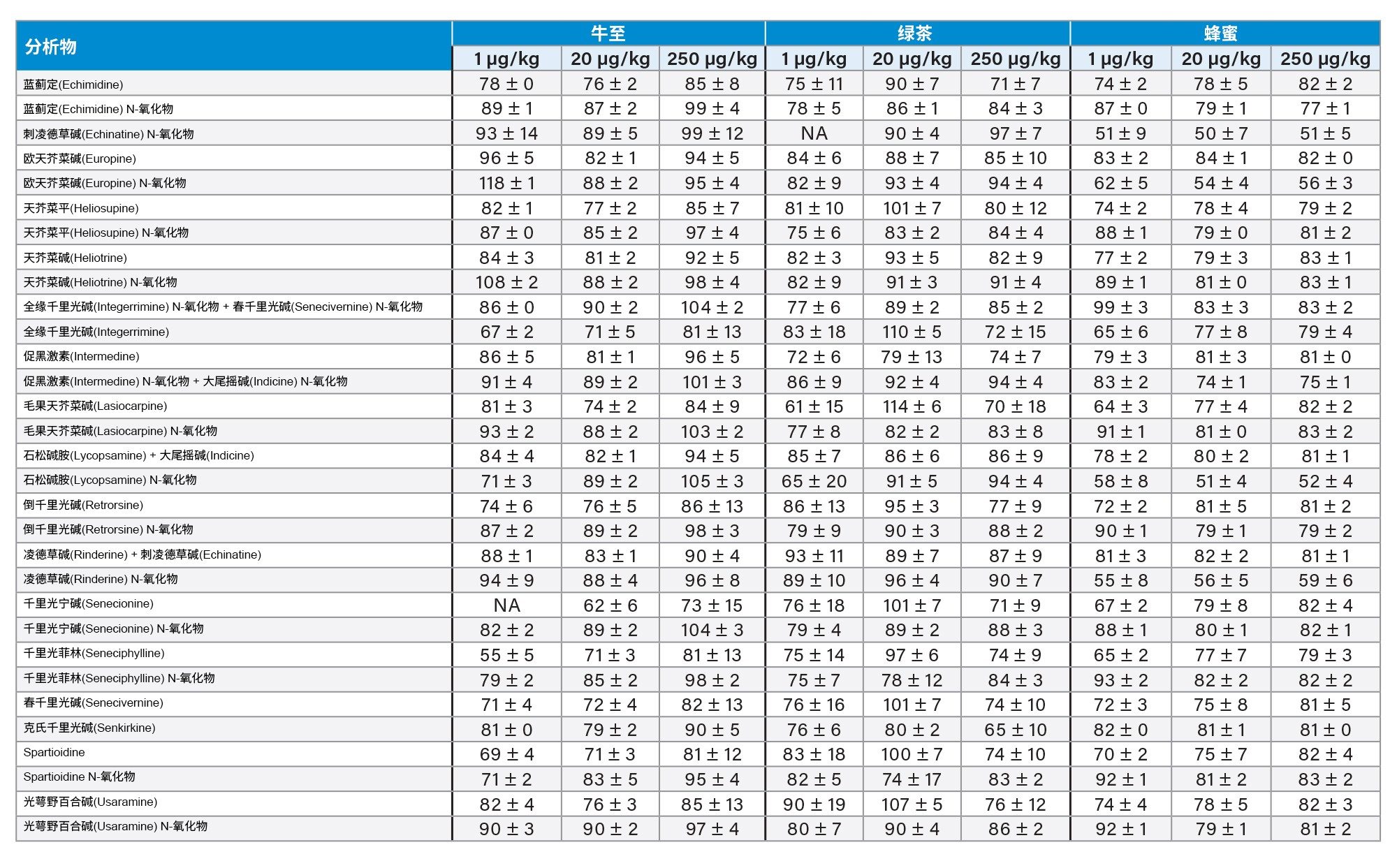 表4.对于法规EU 2020/2040中列出的四种食品以及蜂蜜,在1 µg/kg (n=3)、20 µg/kg (n=6)、250 µg/kg (n=3)加标浓度下,PA的回收率百分比±RSDr%。
表4.对于法规EU 2020/2040中列出的四种食品以及蜂蜜,在1 µg/kg (n=3)、20 µg/kg (n=6)、250 µg/kg (n=3)加标浓度下,PA的回收率百分比±RSDr%。
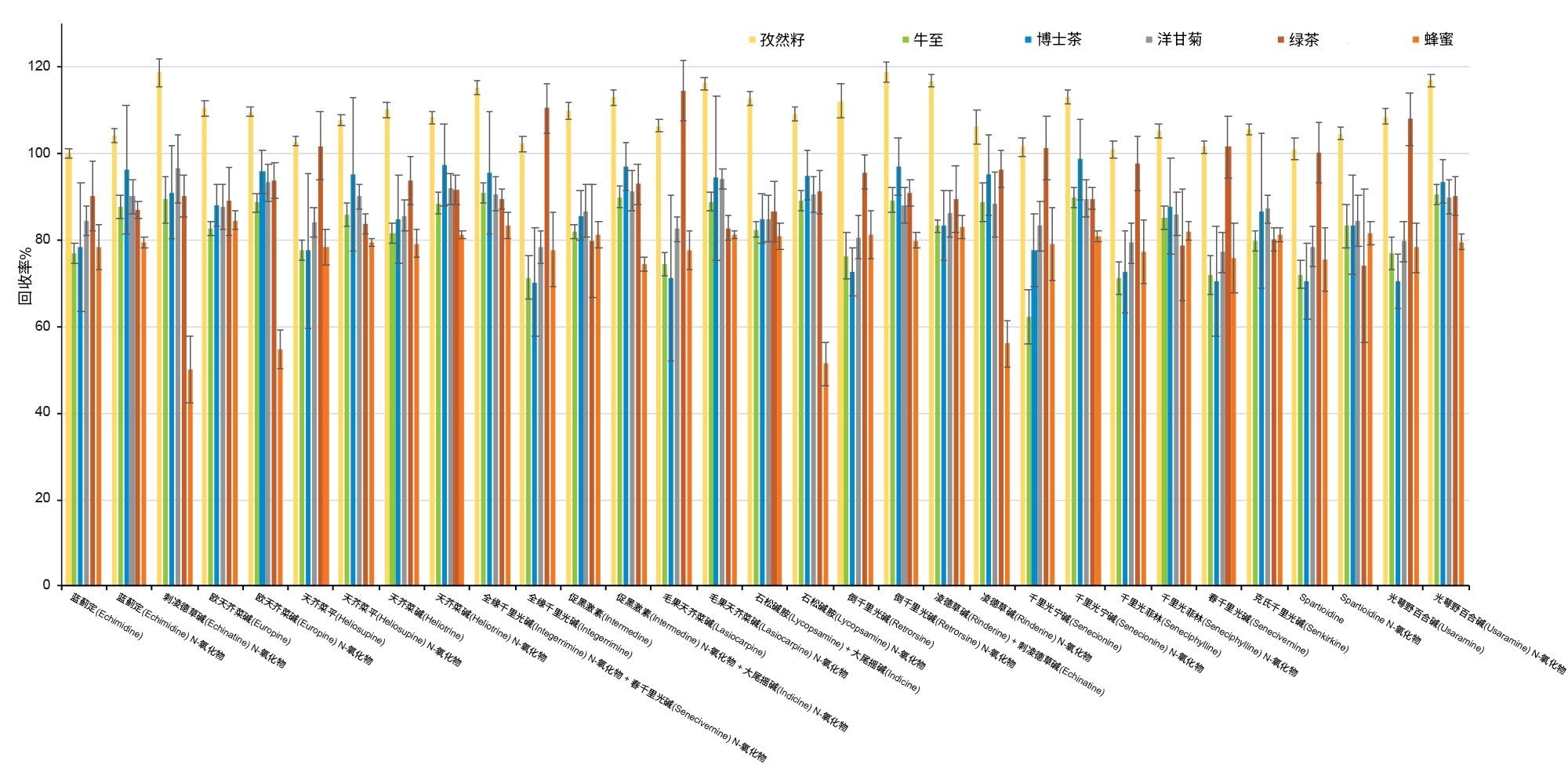 图5.不同食品中加标20 µg/kg标准品获得的回收率百分比条形图(误差条 = %RSDr,n = 6)
图5.不同食品中加标20 µg/kg标准品获得的回收率百分比条形图(误差条 = %RSDr,n = 6)
纯化和基质效应的影响
在大多数情况下,对于草药和香料等植物性样品,由于存在高浓度的污染物,因此需要在萃取后进行一定程度的纯化。本研究利用Oasis MCX SPE消除了一部分可能影响方法性能并污染系统的基质共提取物。本研究采用的SPE方案基于典型的上样-清洗-洗脱方法,利用了酸性条件下质子化碱性化合物(即生物碱)的阳离子交换机制。完成清洗步骤后用碱性溶剂进行洗脱,对于从小柱中选择性回收所有PA至关重要。
为说明SPE纯化的优势,我们利用在浓缩和复溶前应用和不应用SPE纯化进行萃取并添加35种PA的茶样品,采集了色谱图。本研究使用RADAR,这是一种同时采集MRM和全扫描MS的采集模式。两种模式之间的切换仅需5 ms即可完成,确保占空比时间尽可能压缩,同时大幅增加峰上的数据点。图6显示了粗提物全扫描色谱图与纯化后全扫描色谱图的叠加图。与粗提物相比,纯化提取物的总离子流(TIC)信号明显下降。由此显著减少了引入LC-MS/MS的基质共提物数量、减少了可能存在的同分异构体干扰物,并减少了系统污染。
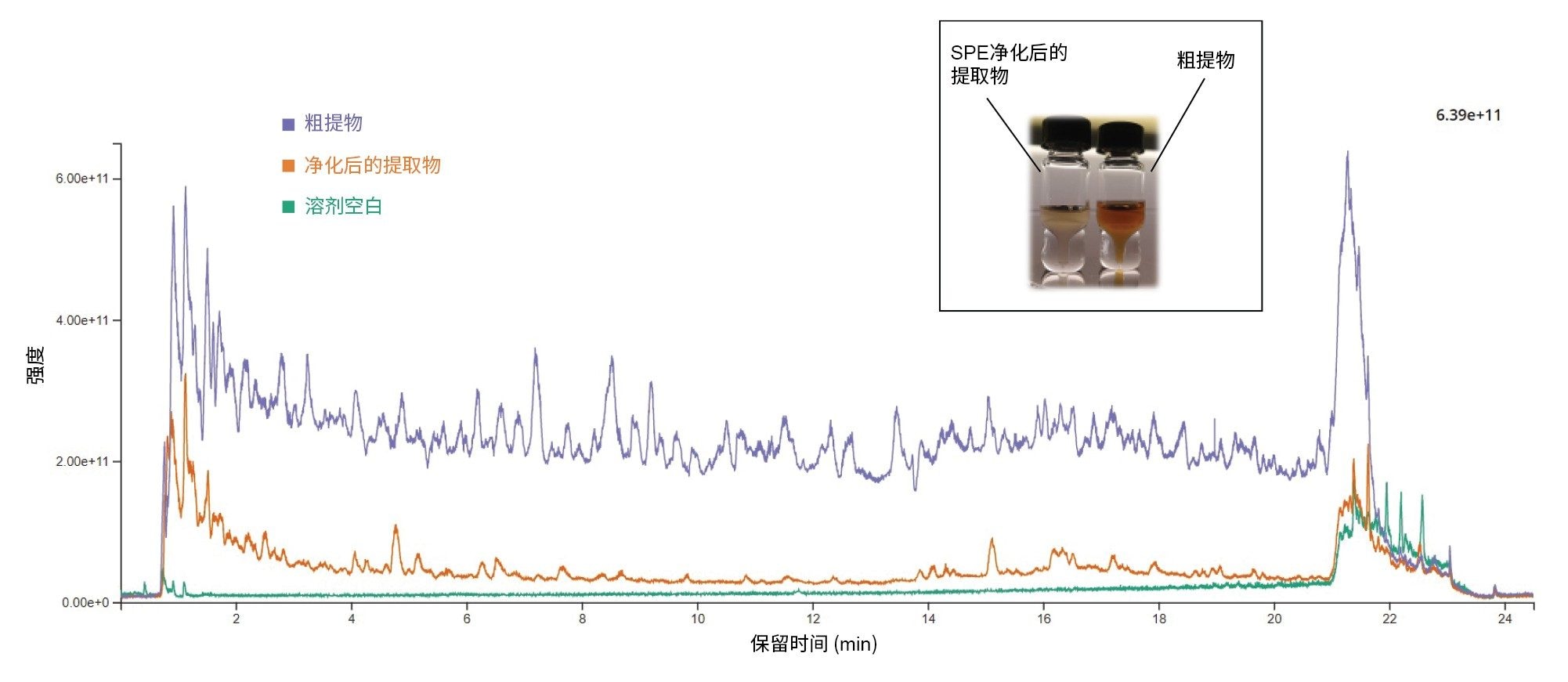 图6.博士茶提取物的RADAR色谱图(全扫描m/z 45~850,扫描时间0.1 s)。插图:纯化提取物(左图)和粗提物(右图)
图6.博士茶提取物的RADAR色谱图(全扫描m/z 45~850,扫描时间0.1 s)。插图:纯化提取物(左图)和粗提物(右图)
复杂基质中PA的准确定量可能会受到ESI中基质效应的影响,导致信号抑制或信号增强。可以预期,在蜂蜜和茶等不同商品以及同一类型的单个样品之间,基质效应程度存在差异。
本文按照下式计算各纯化提取物中的净基质效应:
%ME = (bM / bS -1) * 100
其中bM和bS分别是基质匹配校准曲线和溶剂校准曲线的斜率。净基质效应在-76%~+282%的范围内(见图7)。负值表示基质引起信号抑制,正值表示基质引起信号增强。由此证明,尽管SPE纯化有积极作用,但是仍然需要使用基质匹配校准。
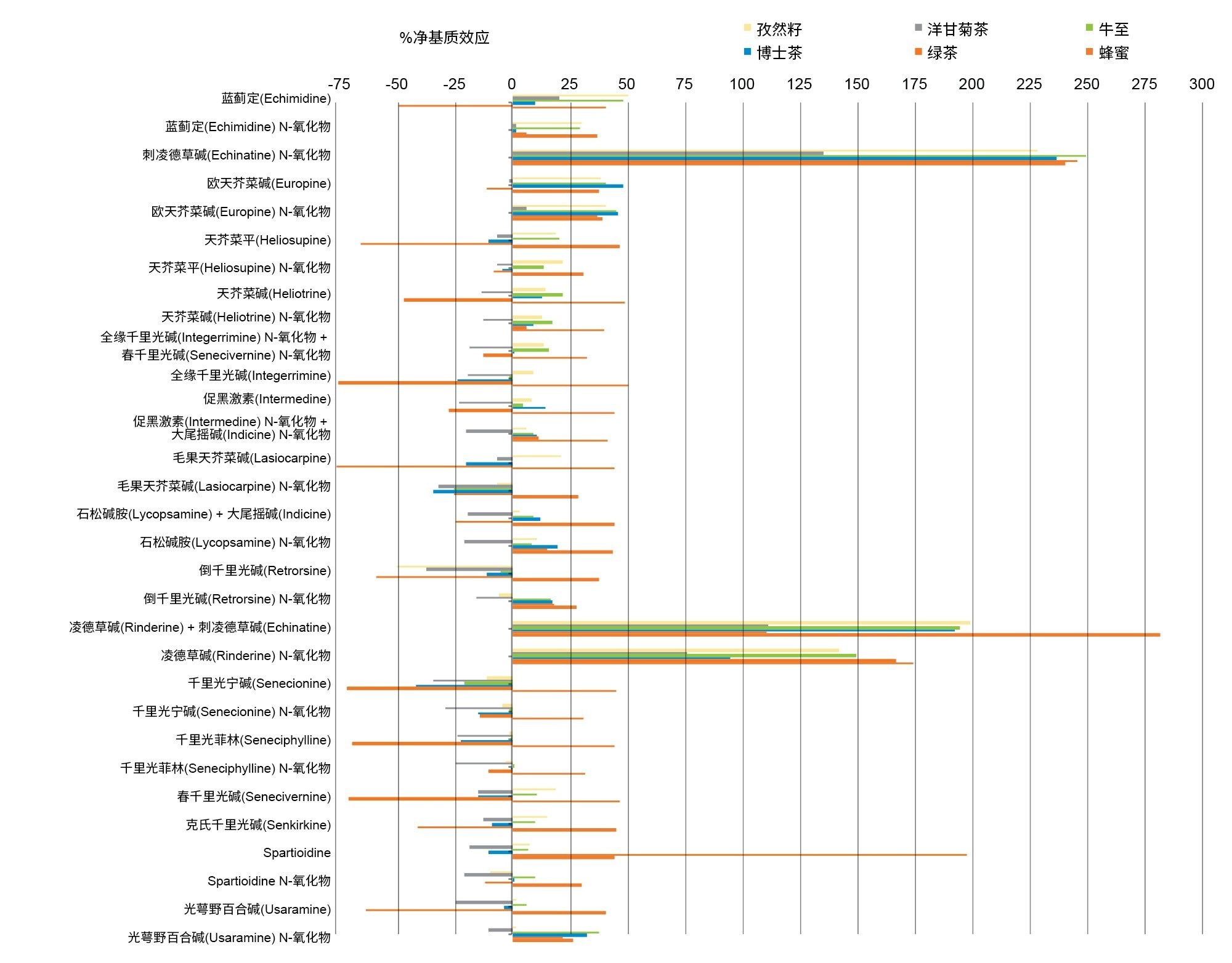 图7.在六种不同类型的样品中计算的每种分析物的净基质效应
图7.在六种不同类型的样品中计算的每种分析物的净基质效应
结论
本研究开发并验证了一种定量测定六种基质(涵盖一系列植物性食品和蜂蜜)中所有35种吡咯里西啶生物碱的合适的UPLC-MS/MS方法。
法规中所列的多种特定异构体使得在常规色谱条件下使用一套全面的方法进行分离非常具有挑战性。在35种分析物中,有27种获得了基线分离,同时我们还定量测定了四对共洗脱异构体的总量,即:石松碱胺(Lycopsamine) + 大尾摇碱(Indicine)、凌德草碱(Rinderine) + 刺凌德草碱(Echinatine)、促黑激素(Intermedine) N-氧化物 + 大尾摇碱(Indicine) N-氧化物和全缘千里光碱(Integerrimine) N-氧化物 + 春千里光碱(Senecivernine) N-氧化物。
研究发现,使用Oasis MCX SPE纯化的优化样品制备程序在减少可能与PA共流出的干扰物质方面非常有效。
我们通过评估正确度、重复性、线性以及检测限和定量限确定了方法性能。结果发现,在考察的所有基质中,LOD/LOQ均显著低于欧盟法规设定的最大限量,因此该方法可用于检测婴幼儿食品,这类产品中PA总量的最大限量为75 µg/kg。
参考资料
- The Plant List, Version 1.1, Released September 2013 (2013).http://www.theplantlist.org.
- Schrenk D., Gao L., Lin G., Mahony C., Mulder P.J. P., Peijnenburg A., Pfuhler S., Rietjens M.C.M. I., Rutz L., Steinhoff B., These A. Pyrrolizidine Alkaloids in Food and Phytomedicine: Occurrence, Exposure, Toxicity, Mechanisms, and Risk Assessment - A Review. Food and Chemical Toxicology 136 (2020) 111107.https://doi.org/10.1016/j.fct.2019.111107.
- EFSA Panel on Contaminants in the Food Chain (Contam); Scientific Opinion on Pyrrolizidine Alkaloids in Food and Feed.EFSA Journal 2011; 9(11):2406.[134 pp.] https://doi.org/10.2903/j.efsa.
- EFSA Panel on Contaminants in the Food Chain (CONTAM); Statement on the Risks for Human Health Related to the Presence of Pyrrolizidine Alkaloids in Honey, Tea, Herbal Infusions and Food Supplements. EFSA Journal 2017; 15(7):4908. [34 pp.] https://doi.org/10.2903/j.efsa.2017.4908.
- Commission Regulation (EU) 2020/2040 of 11 December 2020 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Pyrrolizidine Alkaloids in Certain Foodstuffs.
- Determination of Pyrrolizidine Alkaloids (PAs) In Plant Material by SPE-LC-MS/MS. BfR-PAs-Tea-2.0/2014. https://www.bfr.bund.de/cm/349/determination-of-pyrrolizidine-alkaloids-pa-in-plant-material.pdf
- Determination of Pyrrolizidine Alkaloids (PAs) In Honey by SPE-LC-MS/MS. BfR-PAs-Honey-1.0/2013. https://www.bfr.bund.de/cm/349/determination-of-pyrrolizidine-alkaloids-pa-in-honey.pdf
- Determination of Pyrrolizidine Alkaloids in Plant-Based Food and Feed Materials, Including (Herbal) Teas, Herbal Food Supplements, Fodder and Feedstuffs by LC-MS/MS. EURL-MP-method_002 (v. 3 – 2019).
- RADAR – Understanding Sample Complexity, Improving Quantitative Data Quality.White Paper, Waters Corporation. 2014. https://www.waters.com/nextgen/global/library/library-details.html?documentid=720005033.
- Kwon Y., Koo Y. and Jeong Y. Determination of Pyrrolizidine Alkaloids in Teas Using Liquid Chromatography–Tandem Mass Spectrometry Combined with Rapid-Easy Extraction.Foods (2021), 10, 2250.https://doi.org/10.3390/foods10102250.
- Jeong S. H., Choi E. Y., Kim J., Lee C., Kang J., Cho S., Ko K.Y. LC-ESI-MS/MS Simultaneous Analysis Method Coupled with Cation-Exchange Solid-Phase Extraction for Determination of Pyrrolizidine Alkaloids on Five Kinds of Herbal Medicines.J AOAC Int. (2021), Dec 11;104(6):1514–1525.https://doi.org/10.1093/jaoacint/qsab098.
- Kowalczyk E. and Kwiatek K. Pyrrolizidine alkaloids in honey: Determination With Liquid Chromatography-Mass Spectrometry Method. Journal of Veterinary Research (2018). 62(2), 173–181.https://doi.org/10.2478/jvetres-2018–0027.
- Standard Recommendation S.R. CEN/TR 16059:2010.Food Analysis - Performance Criteria for Single Laboratory Validated Methods of Analysis for the Determination of Mycotoxins.(2010) ICS 07.100.30. European Committee for Standardization.
- Magnusson B. and Örnemark U. (eds.)Eurachem Guide: The Fitness for Purpose of Analytical Methods- A Laboratory Guide to Method Validation and Related Topics, (2nd ed.2014).ISBN 978-91-87461-59-0.
720007624ZH,2022年5月