提高JECFA甜菊糖苷分析方法的色谱分离度
摘要
甜菊糖苷通常作为一种天然的无热量甜味剂用于食品和饮料。这类化合物具有相同的甜菊糖苷配基结构,但糖苷单元的数量和类型不同(例如葡萄糖、鼠李糖或木糖)。FAO/WHO JECFA专论26 (2021)中收录了有关甜菊糖苷的新国际标准,其中推荐使用反相液相色谱法(RPLC)测定主要和次要甜菊糖苷。但是,采用JECFA推荐条件分析甜菊糖苷获得的色谱分离度并不理想(关键分析物对的分离度约为1.0)。本研究在Arc™ Premier系统上使用2.5 µm粒径色谱柱改进了JECFA RPLC方法,所得分离度≥ 1.5,使用亚2 μm粒径色谱柱时,分离度更是提高至2.0。方法优化谨遵ICH Q14“分析方法开发”指导原则中的要素进行。结果表明,改进后的方法准确、灵敏、可靠且稳健,为市售甜叶菊提取物的分析提供了一种更高效的分离方法。
优势
- 遵循ICH Q14“分析方法开发”指导原则中推荐的强化方法优化方案优化了甜菊糖苷的LC-UV分离方法
- 关键分析物对达到了更高的分离度,对复杂甜叶菊提取物的分析很有帮助
简介
甜菊糖苷(SG)是Stevia rebaudiana Bertoni(甜叶菊)这种植物叶片中的成分,其甜味是蔗糖的100至300倍。它们通常作为一种无热量甜味剂用于食品和饮料。目前已经鉴定出的甜菊糖苷有40多种1, 其中丰度最高的是莱苞迪甙A (Reb A)和甜菊苷(SV)。然而,一些次要SG变得更易获得,并且由于甜味更强且苦味回味更少而更受青睐1,2。
液相色谱(LC)是SG分析中使用的主要技术,而反相(RP) C18色谱柱是SG液相色谱分析中相当常用的色谱柱3-6。联合国粮农组织和世界卫生组织(FAO/WHO)下的食品添加剂联合专家委员会(JECFA)自2006年以来发布了一系列有关SG的专论。通过FAO/WHO JECFA专论26 (2021)发表的新专论推荐了两种测定主要SG和次要SG的方法,分别是LC-UV法和LC-UV-MS法1。这两种方法使用相同的C18色谱柱和相同的梯度洗脱条件分离主要SG(13种化合物)和次要SG(17种化合物及其同分异构体),总运行时间为35 min。两种方法的主要区别在于,分析次要SG时使用质谱仪(MS),而分析主要SG时使用紫外/可见光(UV/Vis)检测器。与之前发布的JECFA方法相比,这些方法的色谱分离度有所提高,但仍然不够理想。这些方法的关键分析物对(Reb A/SV)分离度估计可以达到约1.0,但人们仍迫切需要更高效的SG分离方法。
本研究的目的是在不延长运行时间的前提下提高RPLC对SG的色谱分离度。为实现这一目标,我们筛选了五款C18色谱柱,优化了洗脱条件,并评估了分析方法的性能。方法开发谨遵人用药品注册技术要求国际协调会(ICH)指导原则Q14(最终版,2023年11月1日)7中针对分析方法开发的强化方案的要素进行。采用研究开发的方法分析了市售甜叶菊提取物。
实验
标准品和样品
包含甜菊苷、甜菊双糖苷(SVB)、杜克甙A (Dul A)、甜茶苷(Rub)以及莱苞迪甙A、B、C、D和F(Reb A-D、F)的甜叶菊JECFA标准品试剂盒购自ChromaDex(美国科罗拉多州朗蒙特)。莱苞迪甙E (Reb E)和异莱苞迪甙A (Iso Reb A)也购自Chrocadex。莱苞迪甙M、N、O和I(Reb M、N、O和I)购自Sigma-Aldrich(美国密苏里州圣路易斯)。图1显示了本研究使用的SG的结构。这些SG包括JECFA方法中的13种主要SG和另外两种SG(即Reb I和Iso Reb A)。我们网购了五种代表性的市售甜叶菊提取物。这些市售甜叶菊提取物的详细信息见表1。
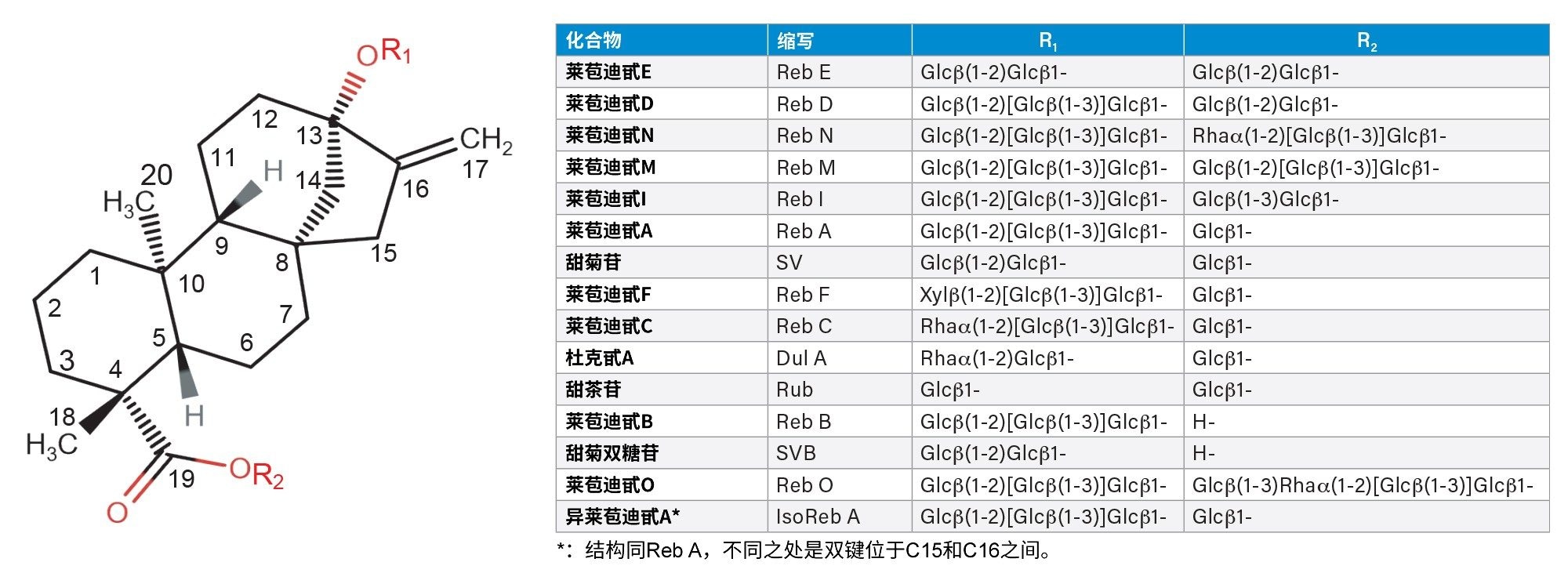 图1.本研究中使用的甜菊糖苷(SG)的结构
图1.本研究中使用的甜菊糖苷(SG)的结构
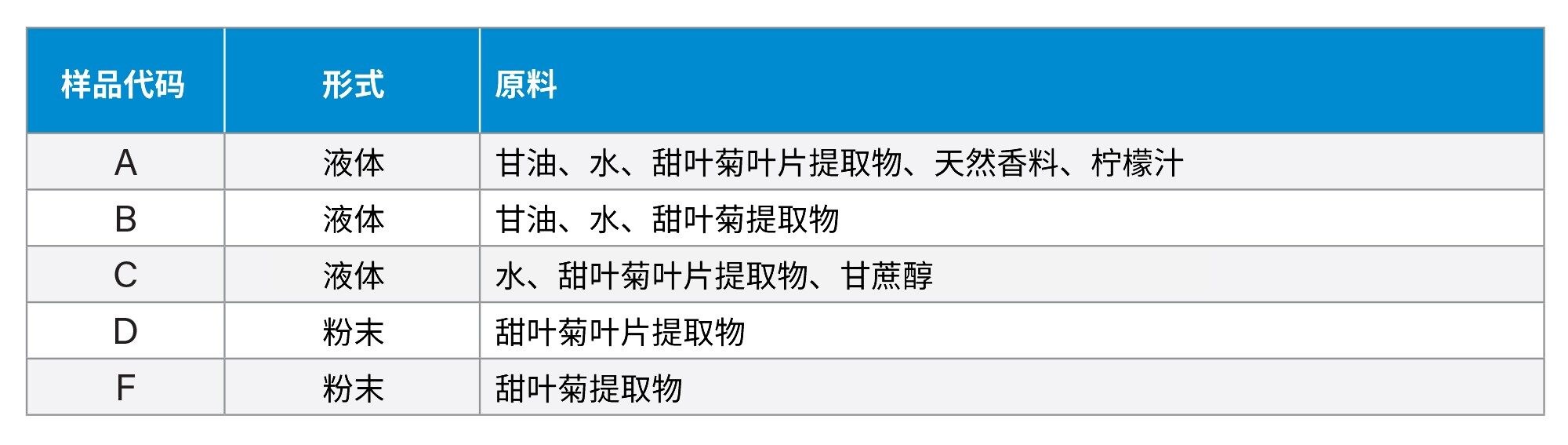 表1.本研究中使用的五种代表性市售甜叶菊提取物的信息
表1.本研究中使用的五种代表性市售甜叶菊提取物的信息
标准品和样品溶液的制备
准确称取5 mg(精确至0.1 mg)标准品,用5 mL稀释剂(乙腈/水混合物,3/7 v/v)稀释,制得单标储备液。混标储备液制备方法如下:移取1.0 mL各单标储备液至20 mL玻璃样品瓶中,在温和的氮气流下挥干,然后用2 mL稀释剂复溶。用不同体积的稀释剂稀释混标储备液,制得浓度为5、20、50、100、200和500 µg/mL的六种工作标准溶液。
精确称取约100 mg样品溶于10 mL稀释剂中,分析前使用0.45 µm PTFE针式过滤器过滤。SG含量高的样品可能需要额外稀释。
方法条件
|
系统: |
配备2998 PDA检测器的Arc Premier系统(BSM) |
|
检测: |
UV (210 nm)和PDA (200~400 nm) |
|
软件: |
Empower™ 3 CDS |
|
流动相A: |
乙腈/水(2:8 v:v,含0.02%甲酸) |
|
流动相B: |
含0.02%甲酸的乙腈 |
|
柱温: |
45 ℃ |
|
样品瓶: |
2 mL螺纹颈口玻璃样品瓶(P/N:186000273),带螺口盖(P/N:186000305) |
使用2.5 μm粒径色谱柱进行分离
|
色谱柱: |
XSelect™ Premier HSS T3 VanGuard™ FIT色谱柱, 130 Å, 2.5 µm, 4.6 mm x 150 mm(P/N:186009863) |
|
进样体积: |
10.0 µL |
|
梯度洗脱程序: |
见表2。 |
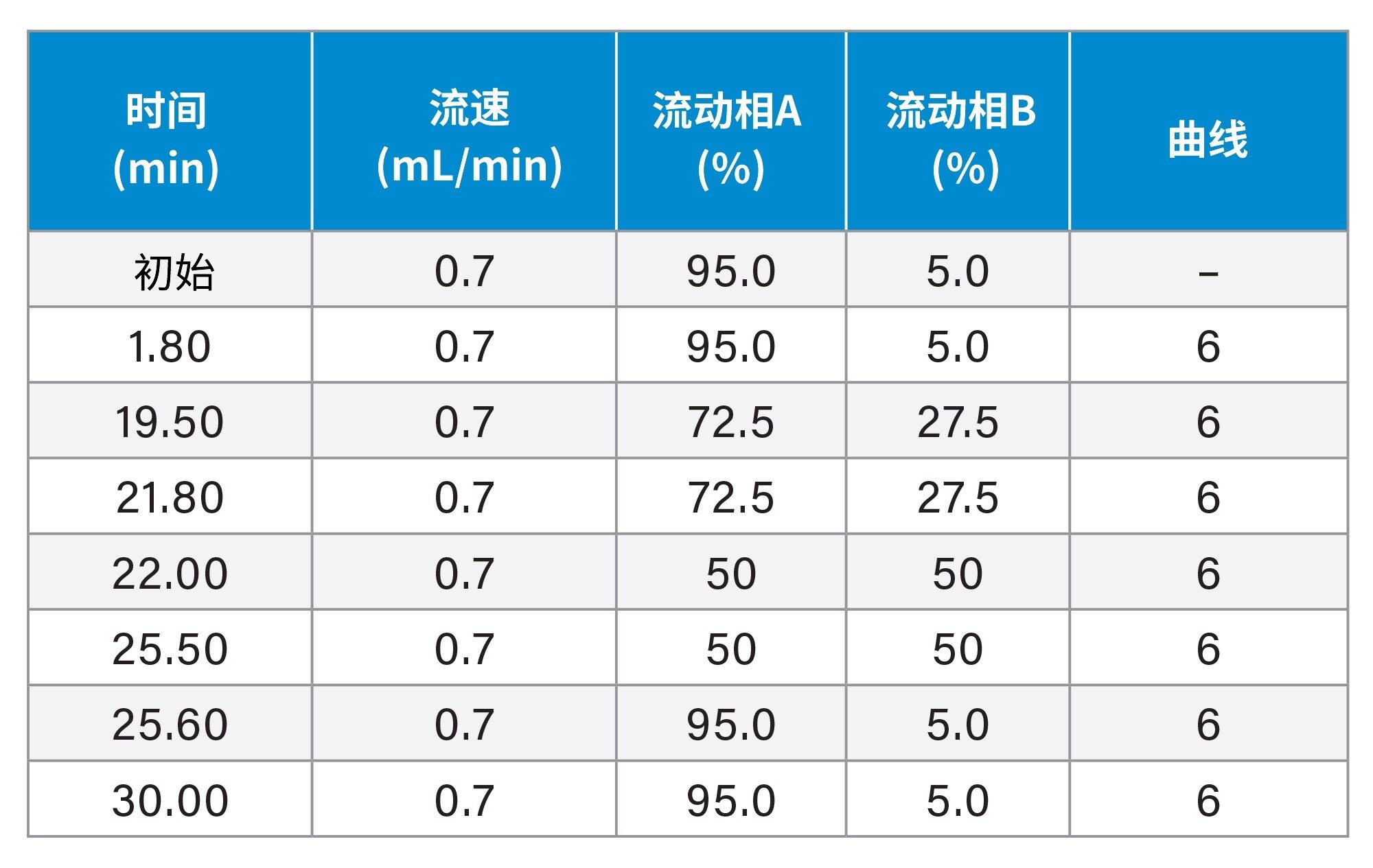 表2. XSelect™ Premier HSS T3 VanGuard FIT色谱柱的梯度洗脱程序
表2. XSelect™ Premier HSS T3 VanGuard FIT色谱柱的梯度洗脱程序
使用亚2 μm粒径色谱柱进行分离
|
色谱柱: |
ACQUITY™ UPLC™ HSS T3色谱柱, 100 Å, 1.8 µm, 3 mm x 150 mm(P/N:186004681) |
|
进样体积: |
5.0 µL |
|
梯度洗脱程序: |
见表3。 |
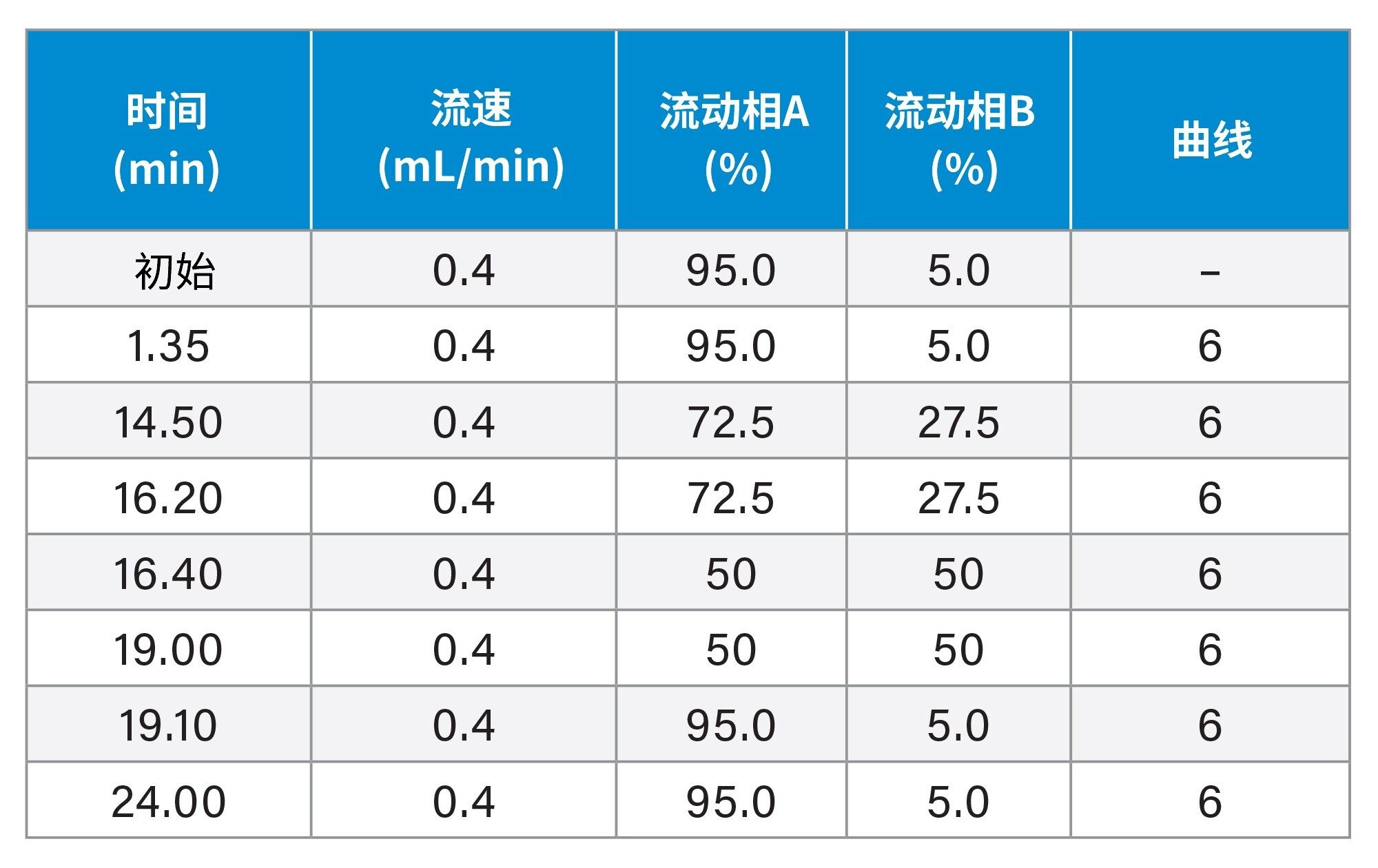 表3. ACQUITY UPLC HSS T3色谱柱的梯度洗脱程序
表3. ACQUITY UPLC HSS T3色谱柱的梯度洗脱程序
方法优化:实验设计
首先采用单变量方案优化梯度洗脱,然后采用多变量方案进行微调。简言之,使用实验设计软件(Fusion QbD®软件,S-Matrix Corp,美国加利福尼亚州尤里卡)优化(单变量优化实验确定的)对分离度影响最大的三个梯度洗脱程序参数,即初始流动相组成、初始保持时间和第一个梯度洗脱的结束时间。具体而言,采用Fusion QbD软件创建基于中心复合面设计的样品队列,然后导出至Empower 3 CDS。在Empower 3 CDS中运行完样品队列并处理好色谱数据后,使用Fusion QbD软件检索结果用于建模和分析。基于Fusion QbD软件的统计分析,得出可接受的性能范围为分离度≥1.70,并确定了理想的色谱条件。我们还借助Fusion QbD软件在理想条件下开展了稳定性测试。(使用Fusion QbD软件)基于全因子设计在离理想条件的2个水平下创建样品队列,用于研究稍稍偏离理想条件对分离度的影响。
结果与讨论
方法开发
我们使用FAO/WHO JECFA专论26推荐的梯度洗脱程序筛选了五款C18色谱柱。筛选的色谱柱列于表4中。所有色谱柱的分离度都相当,但XSelect Premier HSS T3色谱柱对关键分析物对(Reb A/SV)的分离度最高。选择XSelect Premier HSS T3色谱柱开展进一步优化分离条件的实验以获得更高的分离度。我们考察的条件包括柱温(30 ℃~45 ℃)、流速(0.7~1.0 mL/min)和梯度洗脱。采用单变量方案研究这些条件。在初步优化实验中,关键分析物对(Reb A/SV)的分离度达到了1.5。
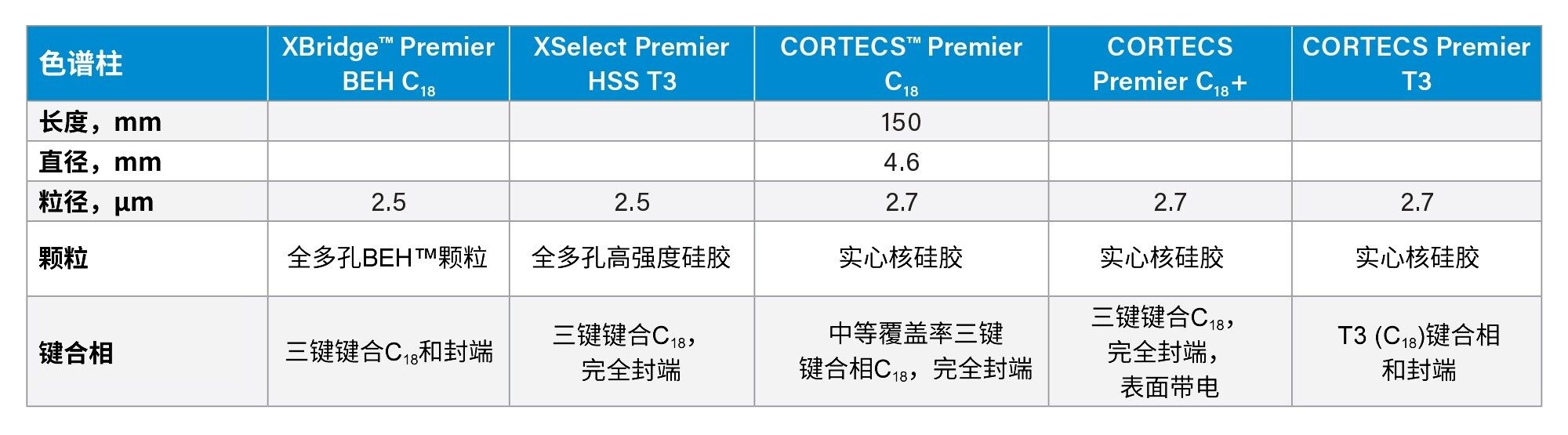 表4.本研究中筛选的色谱柱的详细信息
表4.本研究中筛选的色谱柱的详细信息
梯度洗脱参数(例如洗脱时间和流动相组成)相互之间的依赖性很高,因此,我们采用多变量方案来微调梯度洗脱条件以获得理想的分离度。ICH Q14指导原则推荐采用多变量方案7。 微调洗脱条件后,关键分析物对的分离度达到了1.7。图2显示了四个相当有挑战性的分析物对在分离度至少达到1.70时的可接受性能区域。此外,我们还遵循ICH Q14指导原则,在接近理想液相色谱条件的条件下开展了稳定性测试。图3显示了稳定性测试结果。关键分析物对Reb A/SV的分离度至少达到了1.50,另外几个棘手的SG对(Reb O/Reb D、Reb E/Reb O和SV/Iso Reb A)的分离度至少达到了1.70。表5展示了稳定性测试使用的偏差范围。
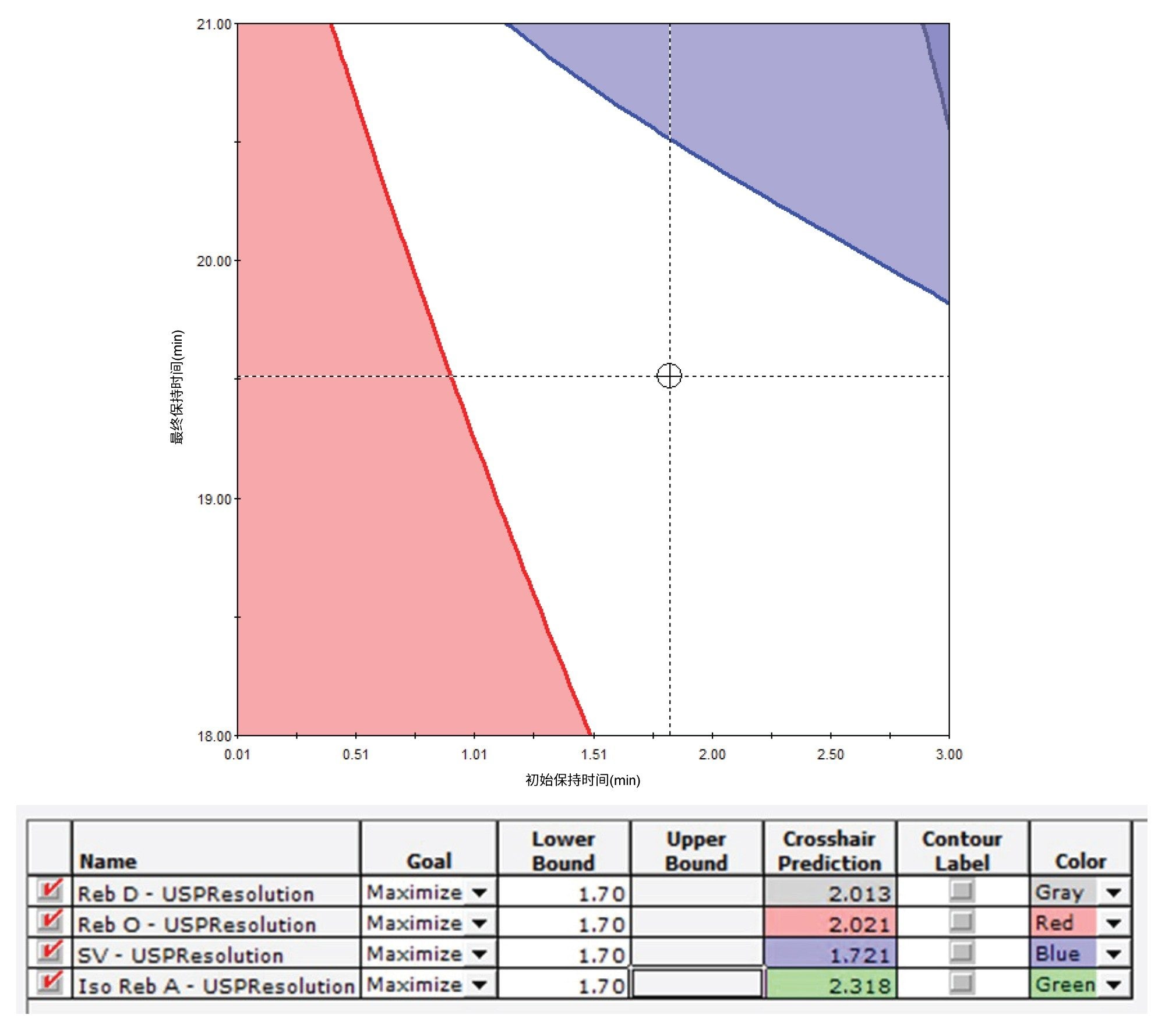 图2.初始流动相组成为5% B时,所得分离度至少达到1.70的可接受性能区域(空白区域)。Reb E/Reb D(显示为Reb D)、Reb D/Reb O(显示为将Reb O)、Reb A/SV(显示为SV)和SV/Iso Reb A(显示为Iso Reb A)这几个关键分析物对被设为优化目标。
图2.初始流动相组成为5% B时,所得分离度至少达到1.70的可接受性能区域(空白区域)。Reb E/Reb D(显示为Reb D)、Reb D/Reb O(显示为将Reb O)、Reb A/SV(显示为SV)和SV/Iso Reb A(显示为Iso Reb A)这几个关键分析物对被设为优化目标。
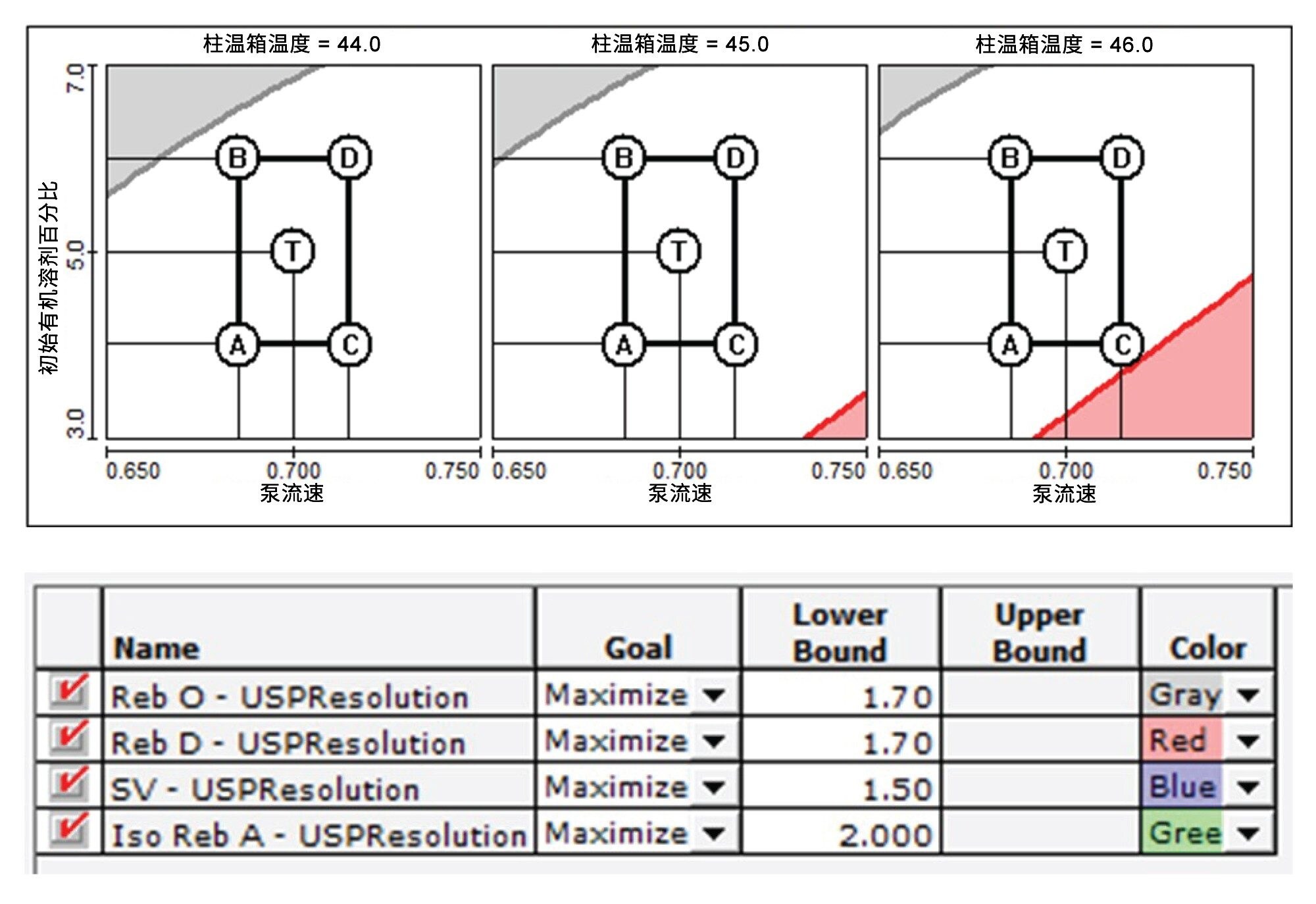 图3.稳定性测试结果。关键分析物对(Reb A/SV)的最低分离度为1.50,其他甜菊糖苷分析物对的最低分离度为1.70。表中的Reb O、Reb D、SV和Iso Reb A分别表示Reb E/Reb O、Reb O/Reb D、Reb A/SV和SV/Iso Reb A这几个分析物对。
图3.稳定性测试结果。关键分析物对(Reb A/SV)的最低分离度为1.50,其他甜菊糖苷分析物对的最低分离度为1.70。表中的Reb O、Reb D、SV和Iso Reb A分别表示Reb E/Reb O、Reb O/Reb D、Reb A/SV和SV/Iso Reb A这几个分析物对。
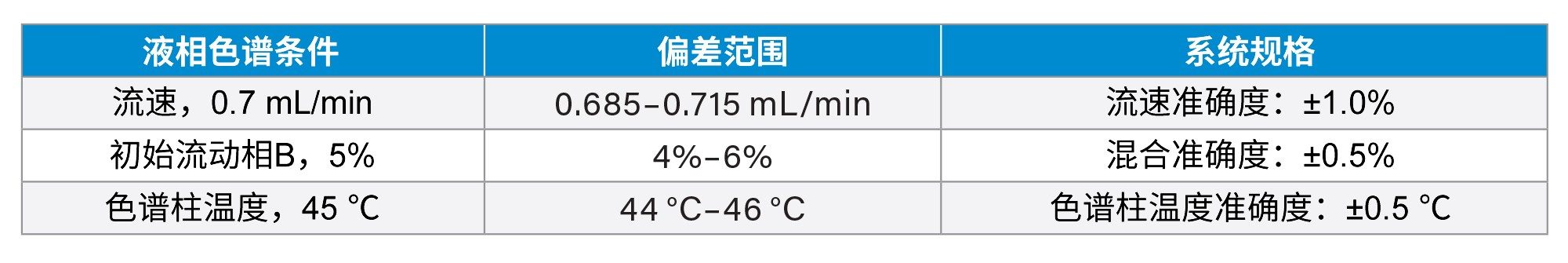 表5.稳定性测试中的液相色谱条件偏差范围和相关系统规格
表5.稳定性测试中的液相色谱条件偏差范围和相关系统规格
方法性能
在Empower 3 CDS中创建PDA数据库,用于存档每个标准样的UV/Vis谱图及其保留时间。根据保留时间和UV/Vis谱图来鉴定峰。此外,使用Empower的峰纯度功能确认峰未发生共洗脱。由于SG的UV/Vis谱图都很相似,因此我们发现保留时间是较为可靠的峰鉴定参数(对于有标准品的SG)。
所有15种SG的峰面积和浓度之间都呈线性关系(表6)。标准曲线采用最小二乘回归模型拟合而得。所有化合物的决定系数(R2)都至少为0.999。将标准曲线上最低浓度水平的响应(峰面积)的标准差(n=5)乘以10,再除以标准曲线的斜率,估算出这些化合物的定量限(LOQ)。估算出的LOQ在0.001~0.004 mg/mL范围内(表6)。
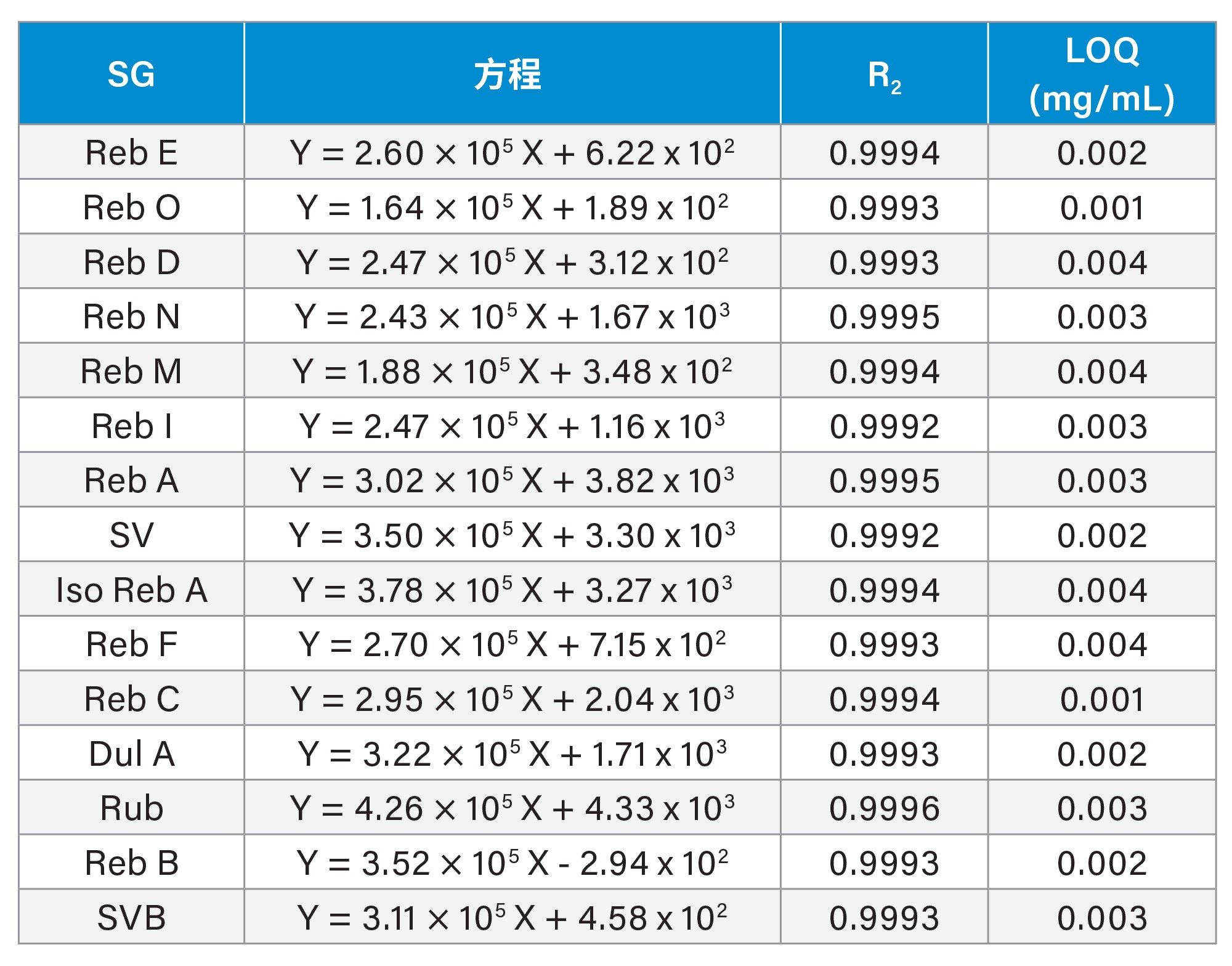 表6.峰面积与浓度之间的线性关系以及估算出的定量限
表6.峰面积与浓度之间的线性关系以及估算出的定量限
通过向形式不同(液体和粉末)且原料不同(样品详细信息见表1)的三个样品中加标两个浓度的SG(Reb N和Reb M)来评估分析准确度。表7显示了加标结果。在大多数情况下,平均回收率(n=2)都在100±10%范围内,只有以低浓度水平加标Reb M的样品B得到了一个例外结果(111.8%)。
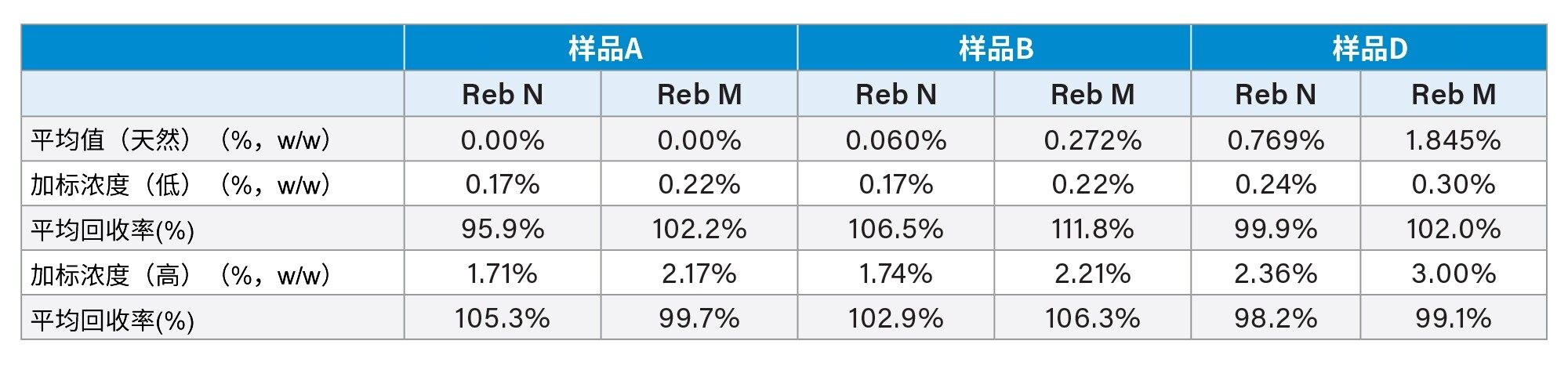 表7.三种样品基质中甜菊糖苷的加标回收率结果
表7.三种样品基质中甜菊糖苷的加标回收率结果
在分析样品时使用QC标准品评估保留时间精密度。15种SG的保留时间相对标准差在0.15%~0.33%范围内(结果未显示)。
如前文所述,在方法开发过程中评估了方法稳定性(请参阅“方法开发”部分)。此外,还在另外两根色谱柱硬件略有不同的HSS T3色谱柱上测试了该方法。其中一根是安装有保护柱(2.5 µm, 3.9 mm x 5 mm)的XSelect Premier HSS T3 VanGuard FIT色谱柱(2.5 µm, 4.6 mm x 150 mm)。另一根是采用常规不锈钢硬件(未采用MaxPeak™高性能表面)的XSelect HSS T3色谱柱(2.5 µm, 4.6 mm x 150 mm)。所有这些色谱柱的填料和色谱柱尺寸基本相同。结果比较见表8。这三根色谱柱(2.5 µm)都获得了优异的分离度,关键分析物对(Reb A/SV)的最低分离度为1.51,其余SG的最低分离度为1.89。采用MaxPeak高性能表面的色谱柱(Premier色谱柱)的分离度优于常规色谱柱(粒径相同),有关MaxPeak高性能表面在该分析中的优势的详细信请参阅其他文章8。 表8还列出了在亚2 μm颗粒色谱柱(ACQUITY UPLC HSS T3色谱柱)上获得的结果。这些结果将在最后一节讨论。
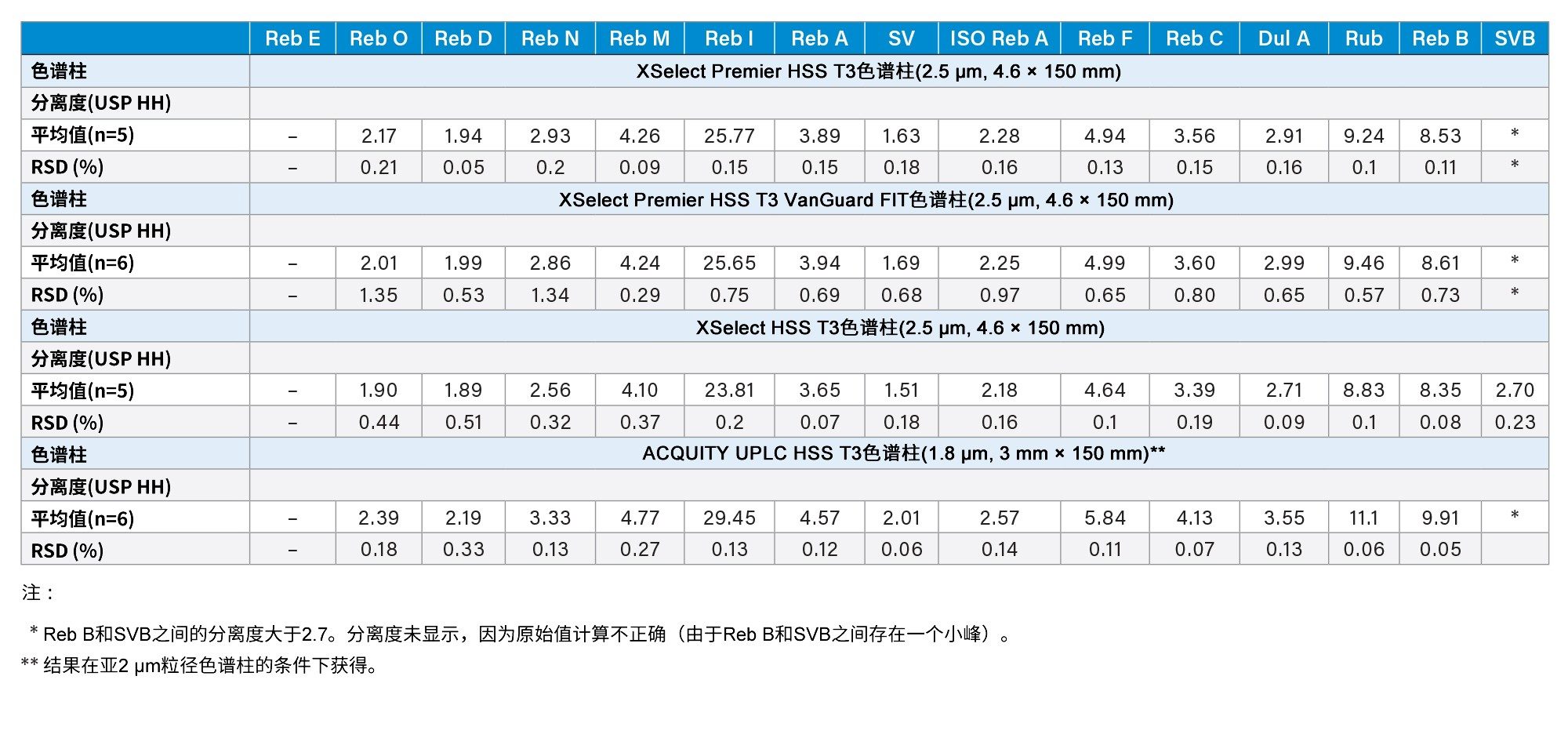 表8.使用不同HSS T3色谱柱对甜菊糖苷进行LC-UV分离的分离度
表8.使用不同HSS T3色谱柱对甜菊糖苷进行LC-UV分离的分离度
样品分析
我们使用实验开发的方法分析了甜叶菊提取物样品(2.5 µm粒径色谱柱的色谱条件见“实验”部分),所得结果如表9所示。混标和样品的HPLC-UV色谱图如图4所示。样品A的SG特征很简单,主要组分是Reb A。其他样品(例如样品B、C、D、F)的SG色谱特征较为复杂。这些样品中存在许多该HPLC-UV方法可能无法鉴定的峰(可能是次要SG)。根据JECFA专论26的建议,在分析SG特征如此复杂的甜叶菊提取物时可能需要使用质谱仪来帮助鉴定化合物。
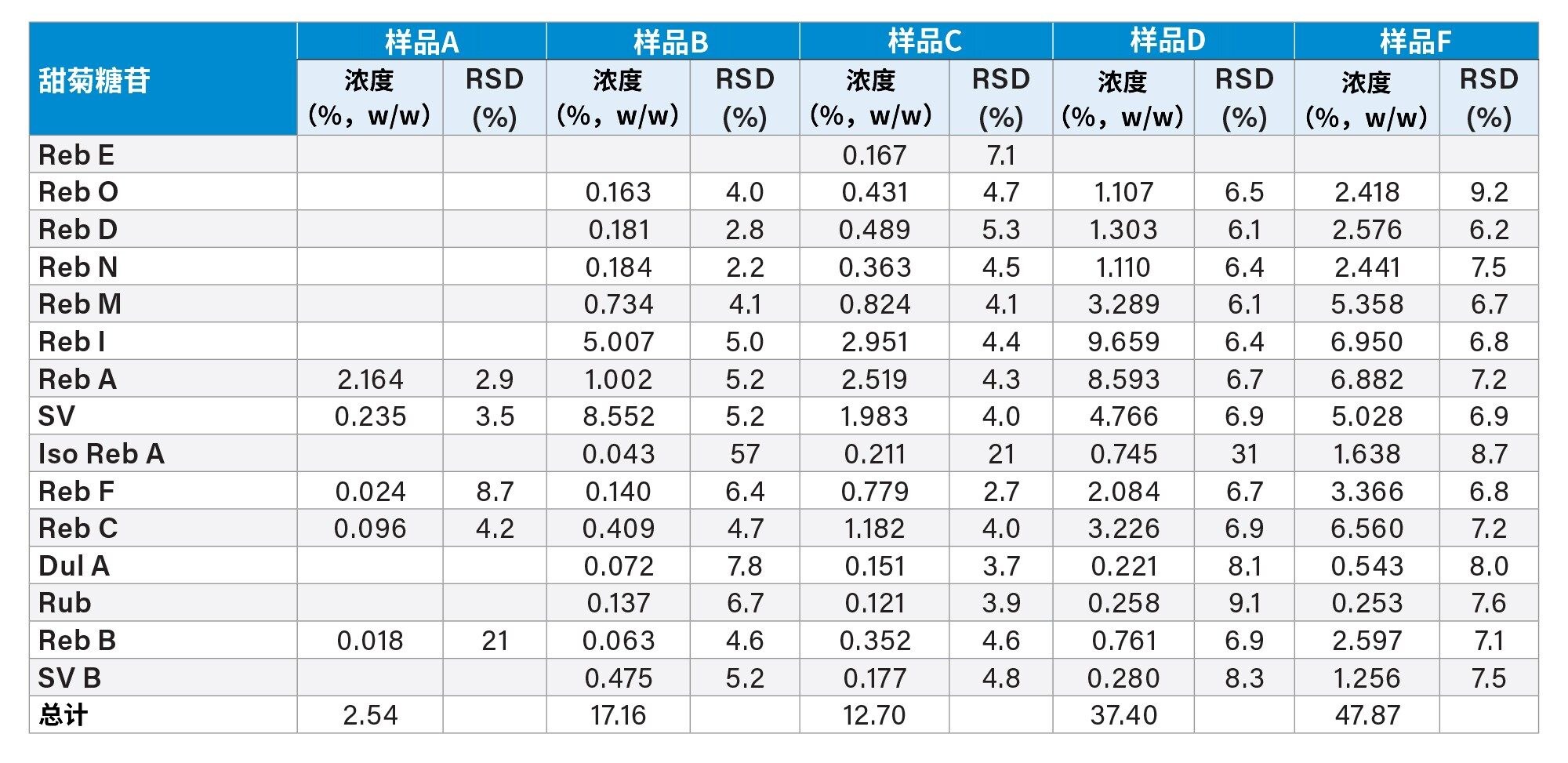 表9.甜叶菊提取物样品分析结果汇总
表9.甜叶菊提取物样品分析结果汇总
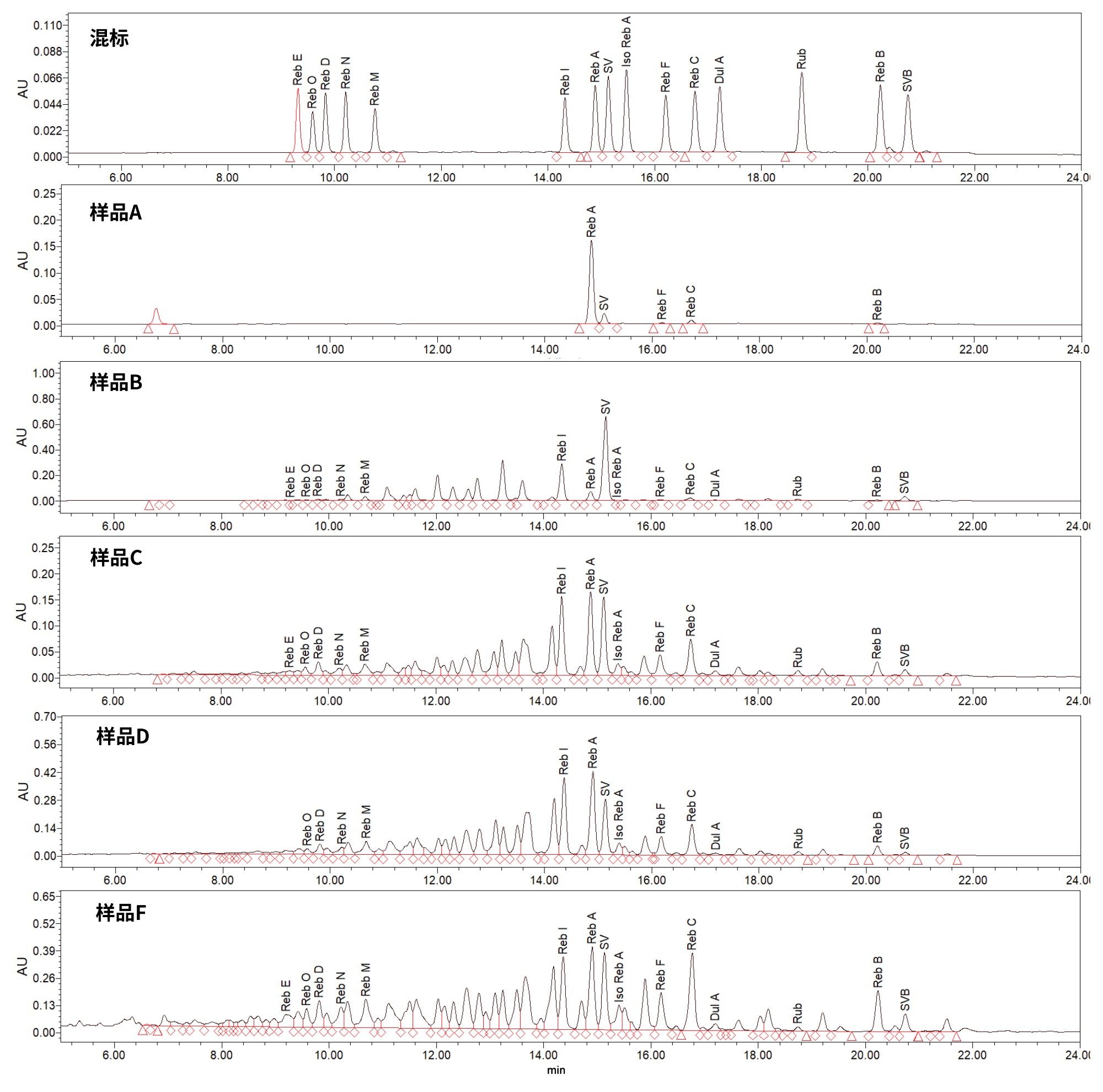 图4.在实验开发的条件下使用Xselect Premier HSS T3 VanGuard FIT色谱柱(2.5 µm, 4.6 mm x 150 mm)分析含15种甜菊糖苷的混标以及甜叶菊提取物产品(样品A、B、C、D、F)得到的HPLC-UV色谱图
图4.在实验开发的条件下使用Xselect Premier HSS T3 VanGuard FIT色谱柱(2.5 µm, 4.6 mm x 150 mm)分析含15种甜菊糖苷的混标以及甜叶菊提取物产品(样品A、B、C、D、F)得到的HPLC-UV色谱图
使用亚2 μm色谱柱进一步提高分离度
如图4所示,甜叶菊提取物的SG特征十分复杂,因此需要进一步提高色谱分离度。为达到更高的分离度,我们将实验开发的方法转换到了具有相同固定相的亚2 μm色谱柱(ACQUITY UPLC HSS T3色谱柱, 1.8 μm, 3 mm x 150 mm)上,并使用方法转换计算器应用程序(2.0版,Waters™ Corp)将梯度洗脱参数转换为适用于1.8 μm粒径色谱柱(3 mm x 150 mm)的新参数(亚2 μm粒径色谱柱和相关色谱条件的详细信息列于“实验”部分)。使用亚2 μm色谱柱时,所有SG对的分离度都达到了至少2.0(见表8)。更小的粒径和更大的柱长/粒径比(L/dp)起到了提高色谱分离度的作用。如果需要在SG分析中达到相当高的色谱分离度,ACQUITY UPLC HSS T3色谱柱也许是更好的选择。请注意,开展本研究时,采用MaxPeak高性能表面的ACQUITY UPLC HSS T3色谱柱尚不可用。
结论
本研究使用配备2998 PDA检测器和XSelect Premier HSS T3色谱柱的Arc Premier系统优化了分析甜菊糖苷的JECFA梯度LC-UV方法,使得SG的色谱分离度显著提高。在XSelect Premier HSS T3色谱柱(2.5 µm, 4.6 mm x 150 mm)上,关键分析物对(Reb A/SV)轻松达到了1.5及更高的分离度,在ACQUITY UPLC HSS T3色谱柱(1.8 µm, 3 mm x 150 mm)上的分离度更是达到了2.0。实验证明,线性、灵敏度、准确度、精密度和稳定性这几方面的分析性能也表现优异。本研究开发的方法有望成为甜菊糖苷分析的一种有用替代方法。
参考资料
- FAO and WHO.2021. Compendium of Food Additive Specifications.Joint FAO/WHO Expert Committee on Food Additives (JECFA), 91st Meeting – Virtual meeting, 1–12 February 2021.FAO JECFA Monographs No.26. Rome.https://doi.org/10.4060/cb4737en.
- Prakash, I.; Markosyan, A.; and Bunders, C. Development of Next Generation Stevia Sweetener: Rebaudioside M. Foods 2014, 3, 162–175; doi: 10.3390/foods3010162.
- Bartholomees, U.; Struyf, T.; Lauwers, O.; Ceunen, S.; Geuns, J. M. C. Validation of an HPLC Method for Direct Measurement of Steviol Equivalents in Foods.Food Chem.2016, 190, 270–275.
- Bergs, D.; Burghoff, B.; Joehnck, M.; Martin, G.; Schembecker, G. Fast and Isocratic HPLC-Method for Steviol Glycosides Analysis from Stevia Rebaudiana Leaves.J. Verbraucherschutz Lebensmittelsicherh.2012, 7, 147–154.
- Zimmermann, B.F. Beaming steviol Glycoside Analysis into the Next Dimension.Food Chem.2018, 241, 150–153.
- Gardana, C.; Scaglianti, M.; and Simonetti, P. Evaluation of steviol and its Glycosides in Stevia Rebaudiana Leaves and Commercial Sweetener by Ultra-High-Performance Liquid Chromatography-Mass Spectrometry.J. Chromatogr.A 1217 (2010) 1463–1470.
- ICH.Analytical Procedure Development Q14, (Final Version, 1 Nov 2023) https://database.ich.org/sites/default/files/ICH_Q14_Guideline_2023_1116.pdf.Accessed 1 January 2024.
- Yang, J; Rainville, P.; Harden, S. Reversed-Phase Liquid Chromatography of Steviol Glycosides - Benefits of MaxPeak High Performance Surfaces.Waters Application Brief, 720008234, 2024.
720008236ZH,2024年2月