药物中N-亚硝基普萘洛尔的LC-MS/MS定量分析
摘要
N-亚硝胺(简称亚硝胺)可能具有潜在的致突变和致癌作用。自2018年以来,亚硝胺已在多个类别的药品中被发现。 相关各方一直致力于减少所有市售药物中的亚硝胺,同时仍确保患者能够安全使用这些重要药物。
在多种药品中已检测到亚硝胺药物相关杂质(NDSRI)。NDRSI的形成是由于药物中存在易受影响的结构特性,例如仲胺或叔胺,这些结构特性在某些化学条件下(包括在辅料中亚硝酸盐的存在)可以引入亚硝基基团。
最近,EMA和FDA都对可接受摄入量(AI)的确定方式进行了修订。这些修订引入了扩展构效关系(SAR)的方法,并根据已知亚硝胺的致癌性数据划分出不同的效力类别(致癌效力分类方法,CPCA)。2022年,普萘洛尔(一种β-受体阻滞剂)因在药品中检测到亚硝基杂质N-亚硝基普萘洛尔而被召回。 本研究介绍了使用超高效液相色谱(UPLC)结合电喷雾检测(ESI)和Xevo™ TQ-S micro串联四极杆质谱仪分析药物中的N-亚硝基普萘洛尔的方法。用于检测NDSRI的分析方法要求其定量下限(LOQ)不超过AI和活性药物成分(API)每日最大剂量(MDD)计算限值的10%。假设MDD为320 mg,则根据N-亚硝基普萘洛尔修订后的AI值(1500 ng/天),该方法的检测性能已优于法规要求。该方法在0.01~100 ppm的N-亚硝基普萘洛尔浓度范围内具有良好的线性关系,浓度偏差小于15%。R2大于0.998。方法回收率范围为89.3%~104.6%,准确度在真值的+/-10%以内。
优势
- 利用Xevo TQ-S micro串联四极杆质谱仪在MRM采集模式实现对N-亚硝基普萘洛尔的痕量检测,性能优于新CPCA方法规定的法规限值要求。
- 该方法准确、重现性良好,用于药物中N-亚硝基普萘洛尔的检测,确保回收率符合规定范围
- 普萘洛尔、N-亚硝基普萘洛尔和N-甲酰基普萘洛尔之间的高效色谱分离
简介
亚硝胺可能具有潜在的致突变和致癌作用1-2。 自2018年以来,亚硝胺已在多个类别的药品中被发现3-5。相关各方一直致力于减少所有市售药物中的亚硝胺,同时仍确保患者能够安全使用这些重要药物6。
亚硝胺药物相关杂质(NDSRI)是一个术语,用于指代活性药物成分(API)与其亚硝基杂质之间的结构关系,这类杂质已在多种药品中检出。NDRSI的形成是由于药物中存在易受影响的结构特性,例如仲胺或叔胺,这些结构特性在某些条件下(包括在辅料中亚硝酸盐的存在)可以引入亚硝基基团2。
最近,EMA和FDA都对可接受摄入量(AI)的确定方式进行了修订7-8。 这些修订引入了扩展构效关系(SAR)的方法,并根据已知亚硝胺的致癌性数据划分出不同的效力类别(致癌效力分类方法,CPCA)。效力得分的计算可以用于分配修订后的AI限值9。目前的效力类别范围从18 ng/天(EMA)、26.5 ng/天(FDA)到1500 ng/天。仪器灵敏度的灵活调节是确保在所有效力类别中准确检测亚硝基杂质的关键。串联四极杆质谱仪分析器具有简单易用、稳定性好、灵敏度高以及分析选择性出色(可在多重反应监测(MRM)实验中选择(采集)母离子和子离子)等优势,为复杂基质(如药品)的定量测定提供了理想的性能特征。
2022年,普萘洛尔(一种广泛用于治疗高血压、心律失常及其他心血管疾病的β-肾上腺素受体阻滞剂(β-受体阻滞剂))因检测到亚硝基杂质N-亚硝基普萘洛尔而被召回,这一检测使用了超高效液相色谱(UPLC™)结合电喷雾检测(ESI)和串联四极杆质谱仪。研究过程中发现,检测的其中一个API样品含有N-甲酰基普萘洛尔(m/z 288),且其碎片离子在不进行色谱分离的情况下,会导致N-亚硝基普萘洛尔的某些MRM通道出现信号重叠。
实验
LC条件
|
液相色谱系统: |
配备FTN的ACQUITY Premier二元系统 |
|
检测: |
PDA 210~400 nm:230 nm单波长 |
|
样品瓶 |
TruView LC/MS(P/N:186005663CV) |
|
色谱柱: |
XBridge™ BEH C8, 2.5 µm, 3.0 × 100 mm(P/N:186006047) |
|
柱温: |
45 ℃ |
|
样品温度: |
5 ℃ |
|
进样体积: |
10 µL |
|
流速: |
0.50 mL/min |
|
流动相A: |
含0.1%甲酸的水溶液 |
|
流动相B: |
含0.1%甲酸的甲醇溶液 |
|
扩充定量环: |
扩充定量环,50 µL,HPS(P/N:700012825) |
质谱条件
|
电离模式: |
电喷雾正离子 |
|
毛细管电压(kV): |
1.0 |
|
锥孔电压(V): |
表2 |
|
碰撞能量(eV): |
表2 |
|
离子源温度(°C): |
130 |
|
脱溶剂气温度(°C): |
500 |
|
脱溶剂气流速(L/h): |
1000 |
|
锥孔气流速(L/h): |
100 |
样品前处理
标准品和试剂
N-亚硝基普萘洛尔确证标准品购自Toronto Research Chemicals(安大略省多伦多)。盐酸普萘洛尔购自Sigma Aldrich(密苏里州圣路易斯)。N-甲酰基普萘洛尔购自BOC Sciences(纽约州Shirley)。Optima LC/MS级溶剂和甲酸购自Fisher Scientific。使用普萘洛尔(1 mg/mL)、N-亚硝基普萘洛尔和N-甲酰基普萘洛尔的初级储备液(10 mg/mL)制备工作溶液。
回收率、准确度和精密度研究
盐酸普萘洛尔(C16H21NO2 · HCl)中普萘洛尔(C16H21NO2)的含量为87.8%。分别称取等份盐酸普萘洛尔(5.7 mg = 5.005 mg普萘洛尔)加入5 mL离心管(Eppendorf)中。用甲醇制备加标溶液,浓度分别为2.5 µg/mL、250 ng/mL和25 ng/mL,置于玻璃闪烁瓶中。这些加标溶液用于制备三个浓度的样品以评估回收率。
预加标样品:移取各浓度的加标溶液10 µL,分别加入6个试管中,每个试管中含有5.7 mg盐酸普萘洛尔。样品平衡30 min后,加入5 mL甲醇,使样品的最终浓度分别为5 ng/mL、0.5 ng/mL和0.05 ng/mL(每个浓度水平n=6)。涡旋混合离心管(1 min)。
后加标样品:称取5.7 mg盐酸普萘洛尔加入5 mL离心管,然后加入5 mL甲醇,制得后加标样品。使用三种加标溶液,分别取10 µL加入甲醇,制得浓度分别为5 ng/mL、0.5 ng/mL和0.05 ng/mL的样品(每个浓度水平n=6)。涡旋混合离心管(1 min)。
基质空白样:向6个装有5.7 mg盐酸普萘洛尔的试管中加入5 mL甲醇,制得基质空白样。涡旋混合离心管(1 min)。
标准品溶液:向5 mL甲醇中加入10 µL加标溶液,在5 mL离心管中制得每个测试浓度水平(5 ng/mL、0.5 ng/mL和0.05 ng/mL,n=6)的标准品溶液。涡旋混合离心管(1 min)。
配制标准曲线标样
确证标准品标准曲线
将含N-亚硝基普萘洛尔确证标准品(10 mg/mL)用甲醇依次稀释,创建0.005~100 ng/mL的标准曲线。
基质标准曲线
称取盐酸普萘洛尔(21.34 mg)加入20 mL玻璃闪烁样品瓶,加入18.7 mL甲醇,制得浓度为1 mg/mL的普萘洛尔储备液。将该溶液用作N-亚硝基普萘洛尔基质标准曲线的稀释剂。使用1 mg/mL普萘洛尔溶液依次稀释N-亚硝基普萘洛尔确证标准品,创建0.005 ppm~100 ppm的标准曲线。
结果与讨论
色谱分离
光电二极管阵列检测器(PDA)数据与MRM数据同步采集。根据观测保留时间(tR),评估了API以及UV检测到的其他杂质与MRM数据中检测到的痕量杂质之间的色谱分离度。为避免MS饱和,在色谱运行的前6.5分钟,使用集成的溶剂切换阀将运行切换至废液。本研究采用C8固定相优化了普萘洛尔API与N-亚硝基普萘洛尔之间的分离(表1和图2)。实现API与痕量杂质的色谱分离非常重要,可以避免由于样品中接近的高浓度API可能引发的基质效应。
MRM通道使用MassLynx™软件的IntelliStart自动调谐功能进行调谐。如果基质引起的噪音增加,多个MRM通道有助于提高特异性。图2展示了其中两个优化的MRM通道,一个是确认离子通道(289>259),另一个是定量离子通道(289>72)。
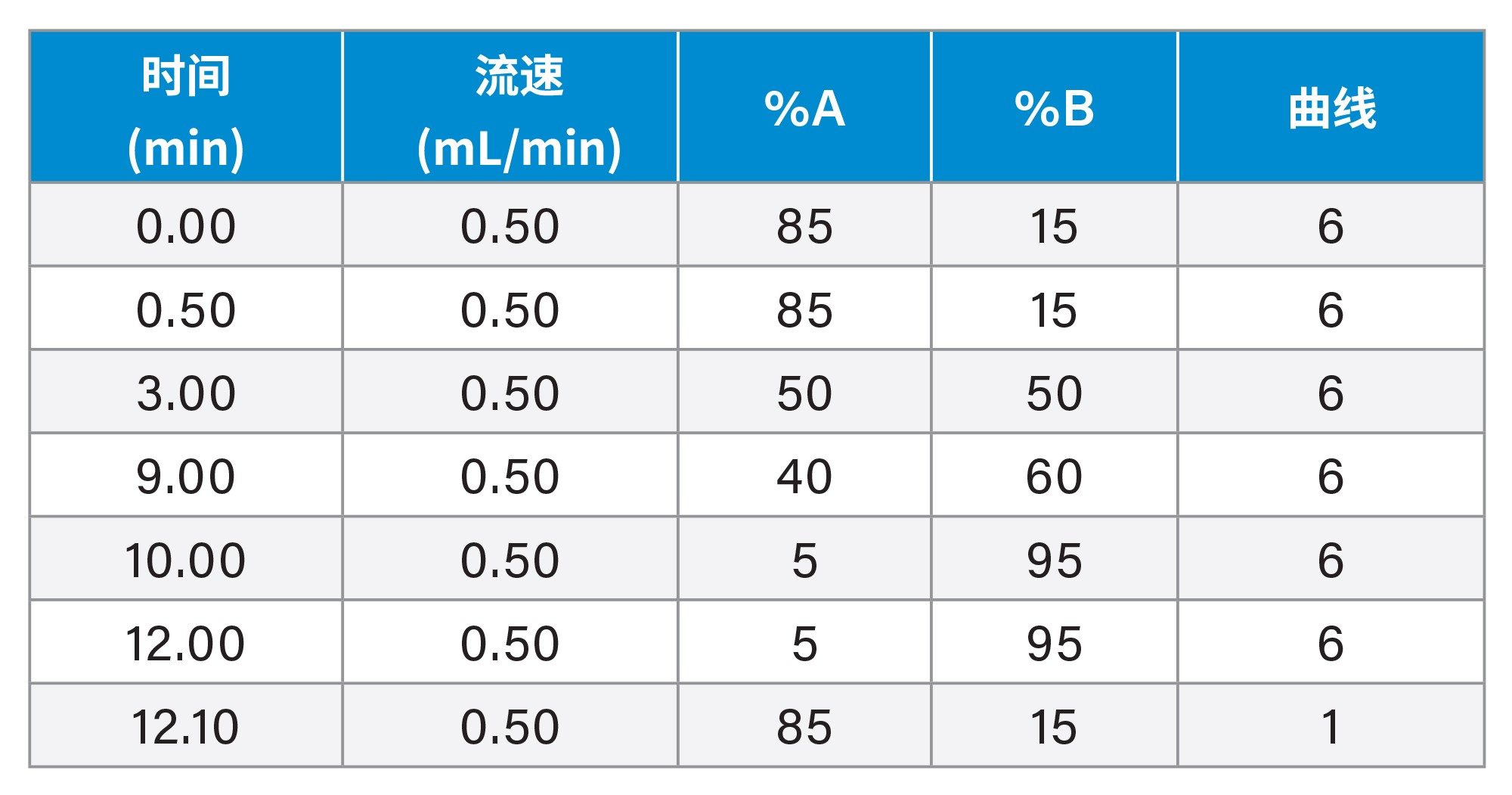 表1.梯度程序。
表1.梯度程序。
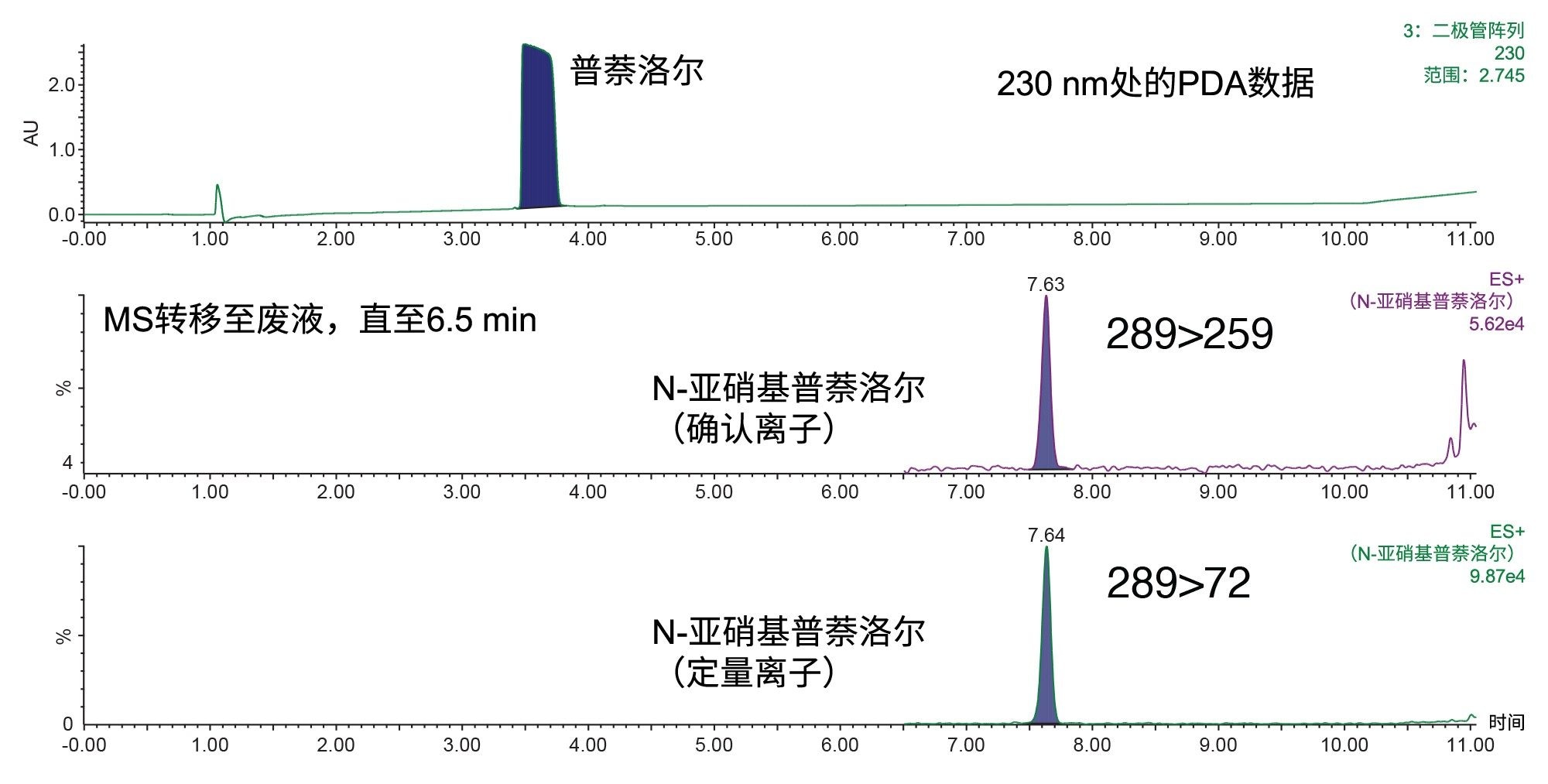 图2. 230 nm下的PDA色谱图(上图),分析对象为1 mg/mL的普萘洛尔API,其中加标了0.1 ppm的N-亚硝基普萘洛尔,进样体积为10 µL,溶剂为甲醇。下方为对应的确认离子和定量离子MRM色谱图。
图2. 230 nm下的PDA色谱图(上图),分析对象为1 mg/mL的普萘洛尔API,其中加标了0.1 ppm的N-亚硝基普萘洛尔,进样体积为10 µL,溶剂为甲醇。下方为对应的确认离子和定量离子MRM色谱图。
N-甲酰基普萘洛尔和N-亚硝基普萘洛尔的分离结果
本研究检测了多种API样品。在其中一个样品中检测到了N-甲酰基普萘洛尔杂质,其结构如图1所示。通过确证标准品验证了N-甲酰基普萘洛尔的鉴定结果。N-甲酰基普萘洛尔的[M+H]+为m/z 288,N-亚硝基普萘洛尔的[M+H]+比其高1 amu,为m/z 289。N-甲酰基普萘洛尔的两个优化MRM通道(288>144和288>102),其母离子和子离子的质量数均比N-亚硝基普萘洛尔的确认离子通道低1 amu(表2)。因此,N-甲酰基普萘洛尔的13C同位素会在N-亚硝基普萘洛尔289>145和289>103的两个MRM通道中产生信号,因为13C的质量数高出1 amu。图3展示了在N-亚硝基普萘洛尔的289>145 MRM通道中与N-甲酰基普萘洛尔相同tR (7.14 min)的色谱峰。因此,为避免这些MRM通道的信号重叠,建议通过色谱进行分离。使用本研究提出的方法可以将N-甲酰基普萘洛尔从N-亚硝基普萘洛尔中分离出来(图3)。
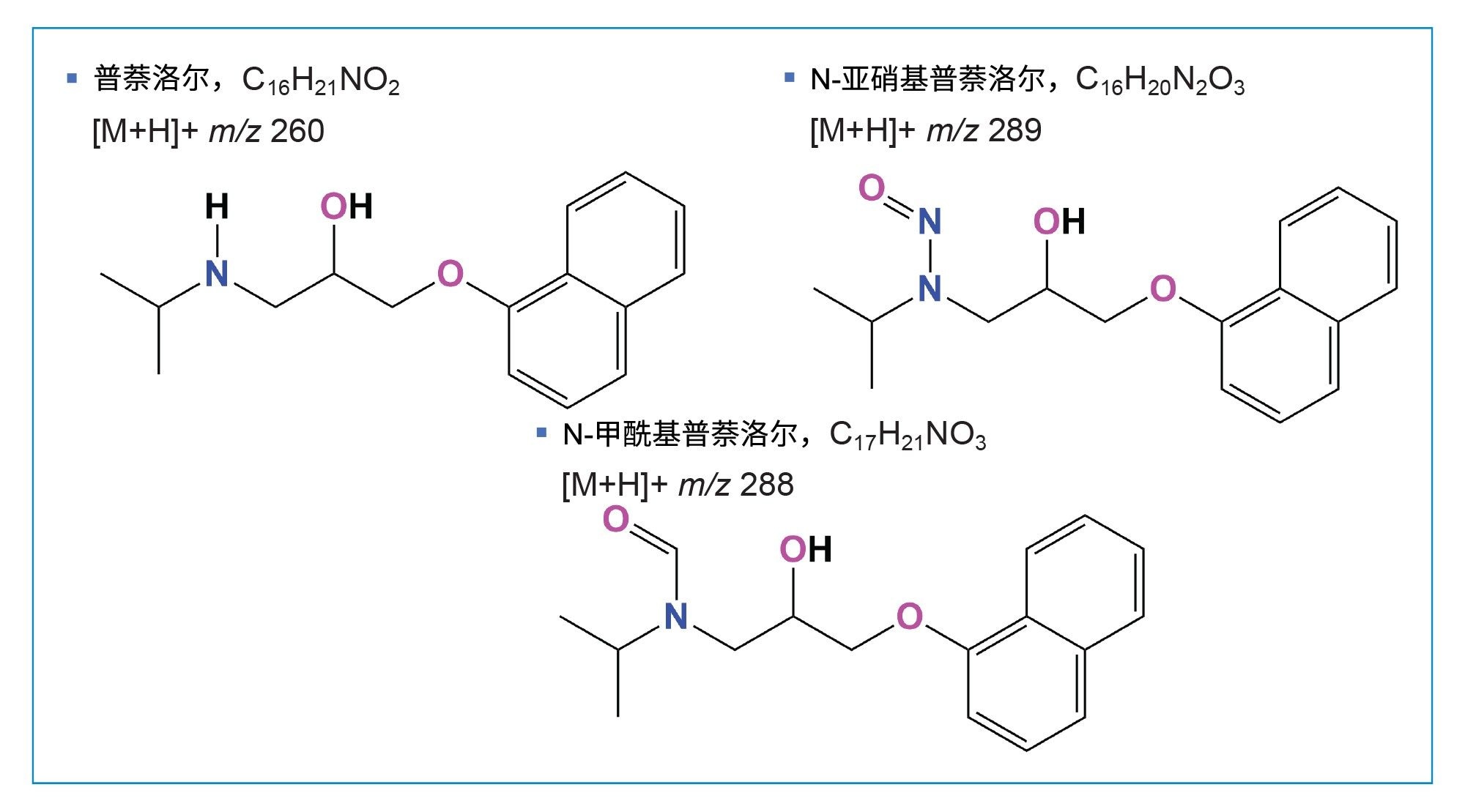 图1.普萘洛尔、N-亚硝基普萘洛尔和N-甲酰基普萘洛尔的结构。
图1.普萘洛尔、N-亚硝基普萘洛尔和N-甲酰基普萘洛尔的结构。
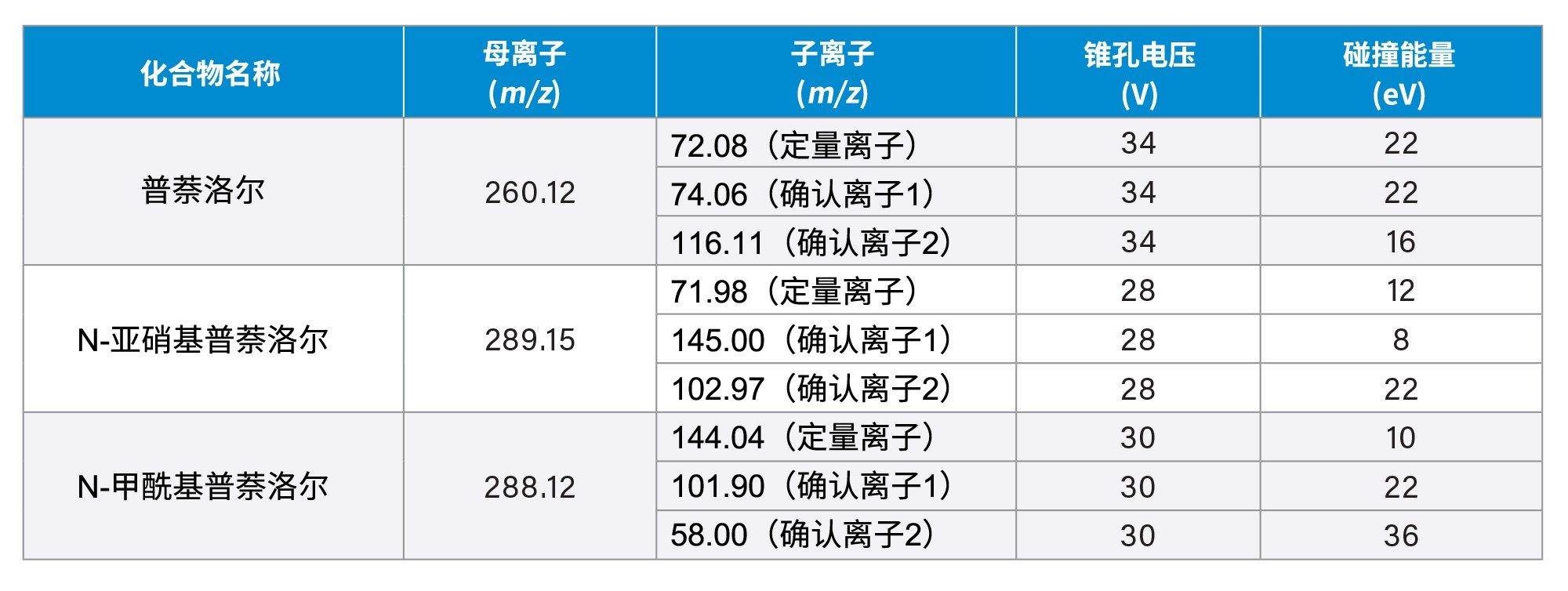 表2.普萘洛尔、N-亚硝基普萘洛尔和N-甲酰基普萘洛尔的定量和确认MRM通道、锥孔电压及碰撞能量设置。
表2.普萘洛尔、N-亚硝基普萘洛尔和N-甲酰基普萘洛尔的定量和确认MRM通道、锥孔电压及碰撞能量设置。
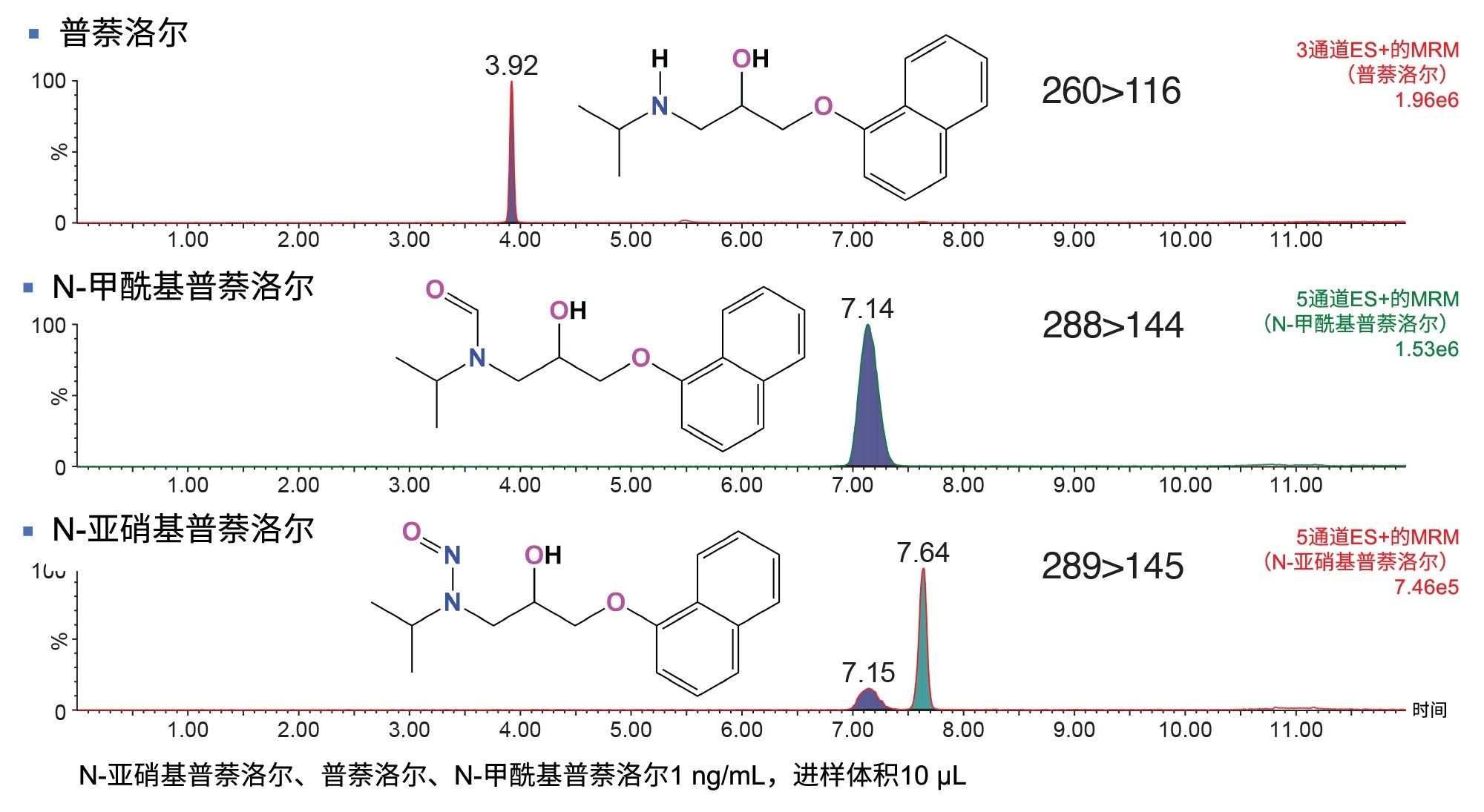 图3.分析普萘洛尔、N-甲酰基普萘洛尔和N-亚硝基普萘洛尔的确证混标(浓度为1 ng/mL,进样体积为10 µL,溶剂为甲醇)所得MRM色谱图。在N-亚硝基普萘洛尔的289>145 MRM通道中,可以看到与N-甲酰基普萘洛尔相同tR的峰。
图3.分析普萘洛尔、N-甲酰基普萘洛尔和N-亚硝基普萘洛尔的确证混标(浓度为1 ng/mL,进样体积为10 µL,溶剂为甲醇)所得MRM色谱图。在N-亚硝基普萘洛尔的289>145 MRM通道中,可以看到与N-甲酰基普萘洛尔相同tR的峰。
色谱峰形
高有机相的进样溶剂可能会导致不良的溶剂效应,从而导致色谱峰形变差。本次分析使用的100%甲醇进样溶液会导致峰形畸变。然而,在流路中增加一定的柱前体积可明显改善峰形。在样品管理器阀的端口6增加一个扩充定量环(体积50 µL,P/N:700012825),可以保持杂质峰形(图4)。
 图4.MRM色谱图,展示了流路中加入(上图)和未加入50 µL扩充定量环(下图)时的峰形比较。普萘洛尔1 mg/mL,其中含有0.1 ppm N-亚硝基普萘洛尔,进样体积为10 µL,溶剂为甲醇。
图4.MRM色谱图,展示了流路中加入(上图)和未加入50 µL扩充定量环(下图)时的峰形比较。普萘洛尔1 mg/mL,其中含有0.1 ppm N-亚硝基普萘洛尔,进样体积为10 µL,溶剂为甲醇。
检测限(LOD)、定量限(LOQ)和线性动态范围
分析的定量限最初是通过N-亚硝基普萘洛尔确证标准品确定的。基于信噪比(S/N) 3:1和10:1分别计算的LOD和LOQ如图5所示。N-亚硝基普萘洛尔确证标准品的LOD和LOQ分别为0.005 ng/mL和0.01 ng/mL。
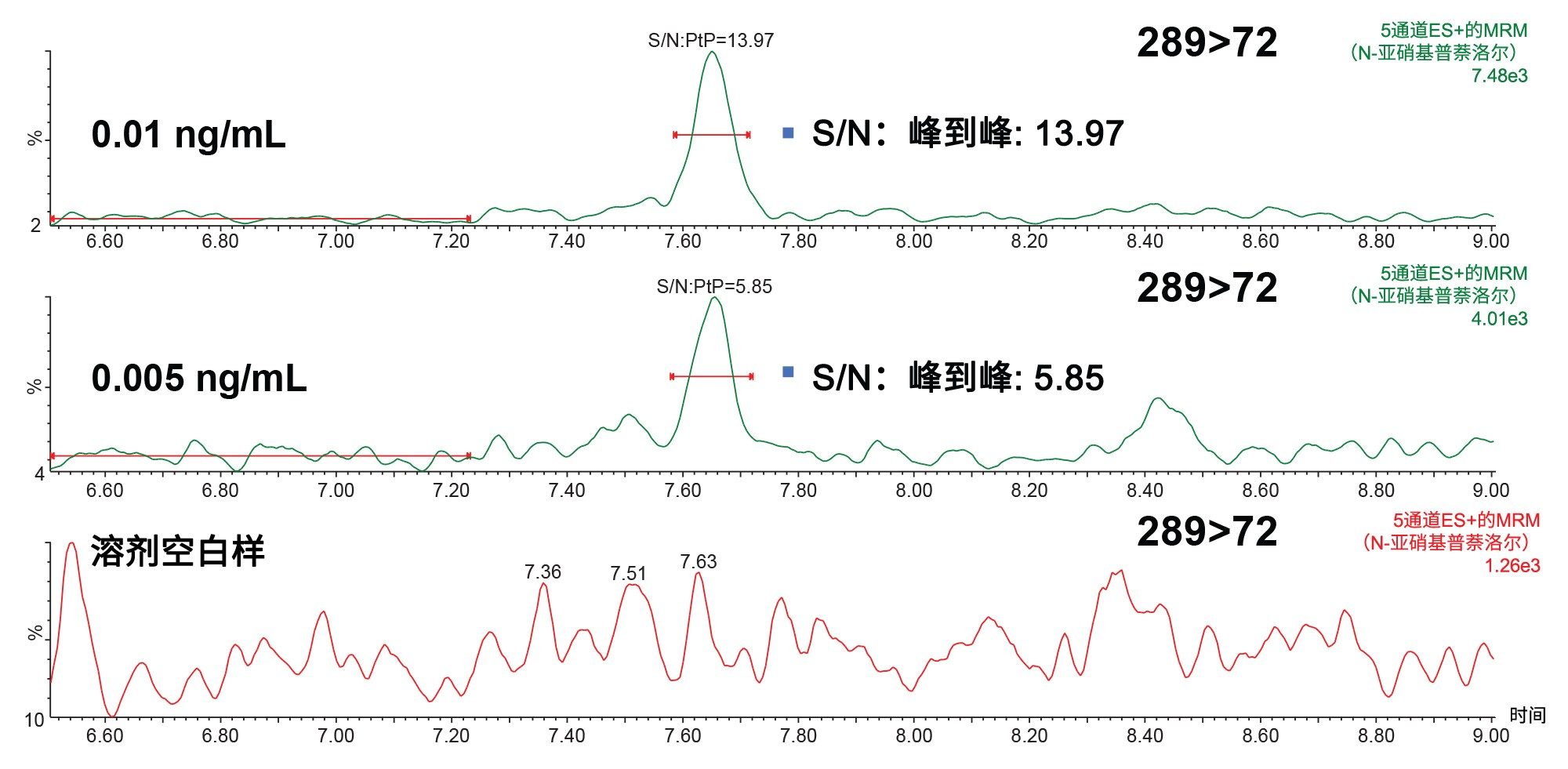 图5. S/N是使用峰到峰(PtP)算法在LOD (0.005 ng/mL)和LOQ (0.01 ng/mL)下测得的,使用N-亚硝基普萘洛尔确证标准品,进样体积为10 µL,溶剂为甲醇。
图5. S/N是使用峰到峰(PtP)算法在LOD (0.005 ng/mL)和LOQ (0.01 ng/mL)下测得的,使用N-亚硝基普萘洛尔确证标准品,进样体积为10 µL,溶剂为甲醇。
确定普萘洛尔API中N-亚硝基普萘洛尔的LOD和LOQ较为复杂,因为每个普萘洛尔API样品中都存在这种内源性杂质。由1 mg/mL普萘洛尔配制的标准曲线的低浓度部分如图6所示。
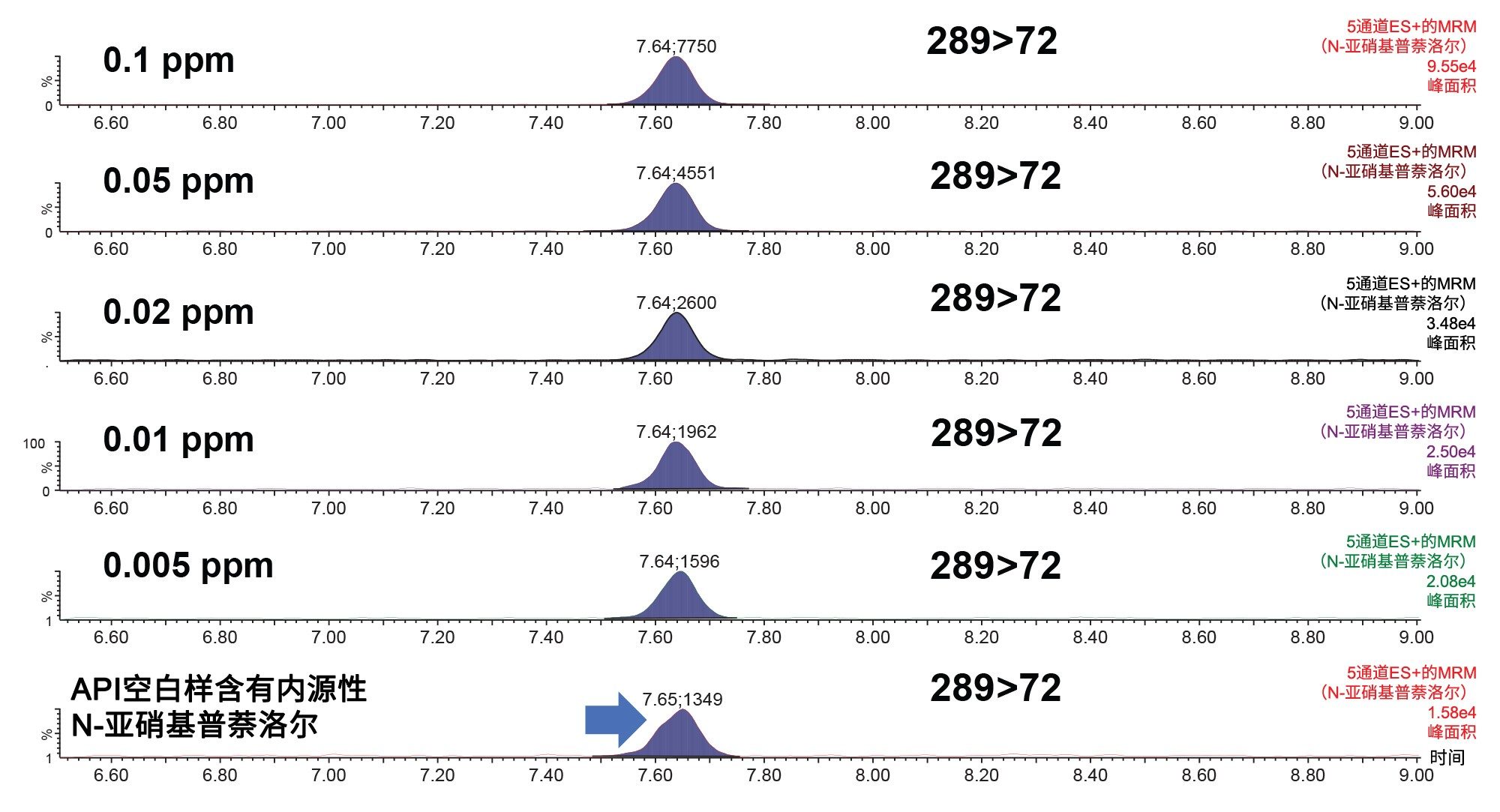 图6. 1 mg/mL普萘洛尔中N-亚硝基普萘洛尔较低标准曲线浓度的分析MRM色谱图。基质空白样(未加标API)中的峰响应。
图6. 1 mg/mL普萘洛尔中N-亚硝基普萘洛尔较低标准曲线浓度的分析MRM色谱图。基质空白样(未加标API)中的峰响应。
表3汇总了加标0.005 ppm和0.01 ppm N-亚硝基普萘洛尔的API样品(1 mg/mL普萘洛尔)6次重复进样获得的平均峰面积响应。此外,还列出了N-亚硝基普萘洛尔确证标准品在LOD 0.005 ng/mL和LOQ 0.01 ng/mL时的平均峰面积响应。使用API本身所含的N-亚硝基普萘洛尔(内源性杂质)的平均峰响应(n=6),对加标API样品中N-亚硝基普萘洛尔的响应进行校准。然后将校正后的加标样品峰面积响应与确证标准品的响应进行比较。结果显示,API中LOD和LOQ浓度下的校正响应与相同浓度确证标准品中观察到的响应相差均在10%以内。
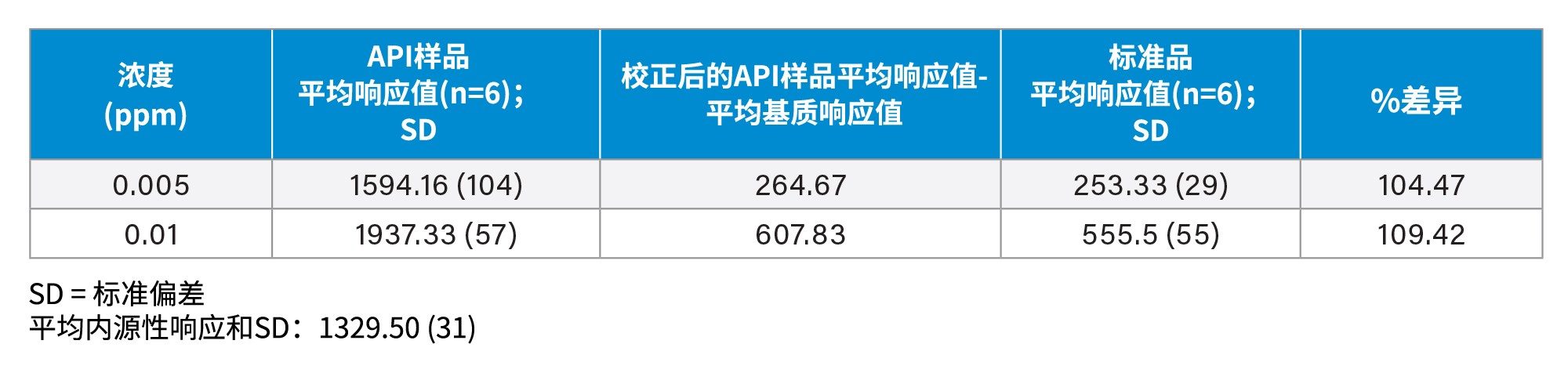 表3.在LOD (0.005 ppm)和LOQ (0.01 ppm)下,加标N-亚硝基普萘洛尔的API样品的峰面积响应、加标样品的校正响应以及确证标准品样品的响应比较。
表3.在LOD (0.005 ppm)和LOQ (0.01 ppm)下,加标N-亚硝基普萘洛尔的API样品的峰面积响应、加标样品的校正响应以及确证标准品样品的响应比较。
N-亚硝基普萘洛尔的阈值水平为4.69 ppm,其阈值水平的10%为0.469 ppm。图7展示了在1 mg/mL普萘洛尔溶液中分别加标5 ppm和0.5 ppm N-亚硝基普萘洛尔的响应、API空白基质的响应结果,以及使用PtP算法计算的S/N,结果表明该方法的灵敏度高于法规阈值水平的要求。
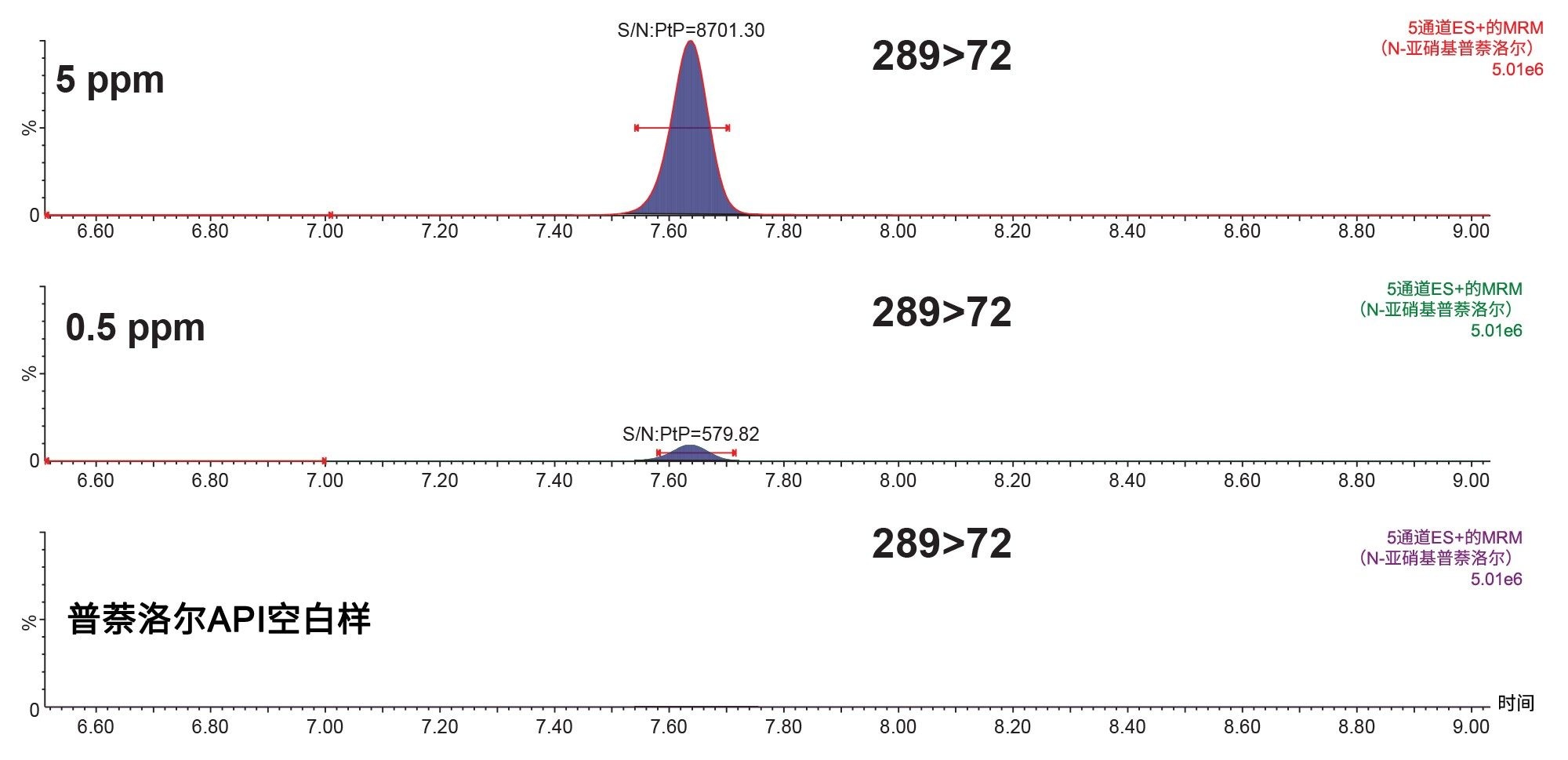 图7. 加标5 ppm和0.5 ppm N-亚硝基普萘洛尔的1 mg/mL普萘洛尔溶液的分析MRM色谱图,同时还展示了API空白样和PtP S/N。
图7. 加标5 ppm和0.5 ppm N-亚硝基普萘洛尔的1 mg/mL普萘洛尔溶液的分析MRM色谱图,同时还展示了API空白样和PtP S/N。
在API存在的情况下评估N-亚硝基普萘洛尔之前,使用N-亚硝基普萘洛尔确证标准品确定线性动态范围。三次进样的曲线在0.005~100 ng/mL范围内表现出良好的线性,R2为0.998,该范围内的浓度偏差小于10%(数据未展示)。
在1 mg/mL API中N-亚硝基普萘洛尔的标准曲线如图8所示。使用1/X加权时,R2为0.998,浓度范围0.01~100 ng/mL(覆盖4个数量级)内的浓度偏差均小于15%。
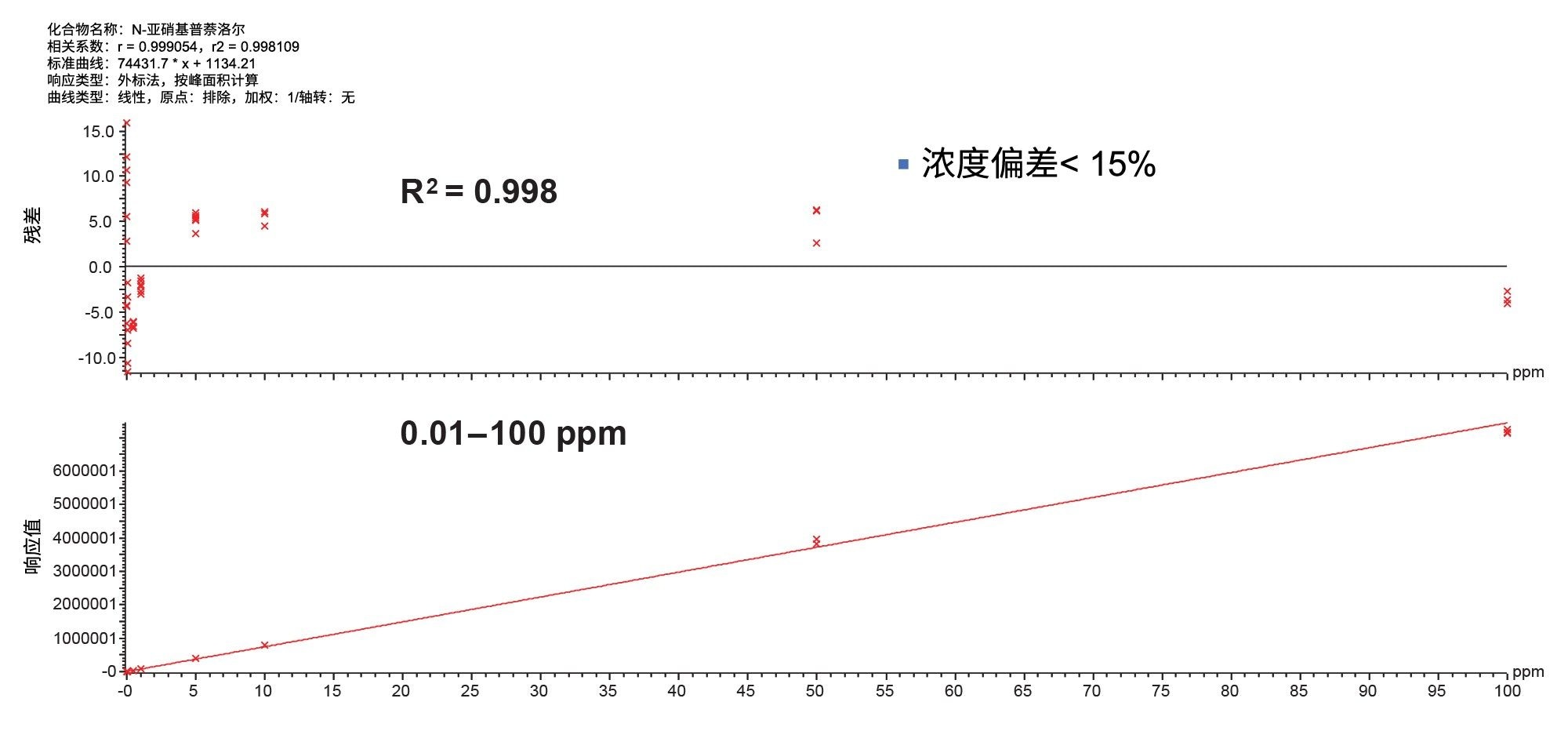 图8.在1 mg/mL的普萘洛尔溶液中,N-亚硝基普萘洛尔的线性范围为0.01~100 ng/mL,在测试的标准曲线范围内残差< 15%,重复进样三次。
图8.在1 mg/mL的普萘洛尔溶液中,N-亚硝基普萘洛尔的线性范围为0.01~100 ng/mL,在测试的标准曲线范围内残差< 15%,重复进样三次。
回收率、精密度和准确度
回收率实验分别在0.05 ppm、0.5 ppm和5 ppm (n=6)下进行。结果汇总见表4。由于API中存在内源性N-亚硝基普萘洛尔的水平,回收率的计算基于预加标API样品的校正响应,校正时使用API空白样(n=6)中N-亚硝基普萘洛尔峰的平均响应值。从预加标样品的响应值中扣除API空白样中该峰的平均响应值。回收率使用以下公式计算:
%回收率 = (经校正的预加标响应值/标准品响应值) * 100%
计算得到的回收率在89.3%~104.6%之间。各浓度水平回收率的%RSD均低于5%。
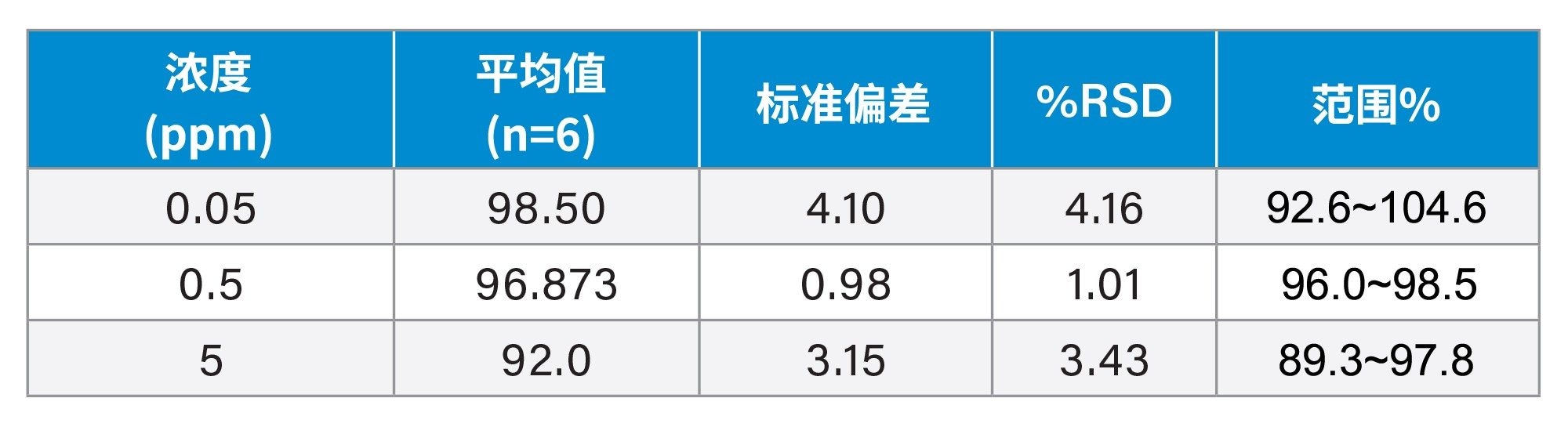 表4.在1 mg/mL API中,加标0.05 ppm、0.5 ppm和5 ppm样品(n=6)的方法回收率汇总。
表4.在1 mg/mL API中,加标0.05 ppm、0.5 ppm和5 ppm样品(n=6)的方法回收率汇总。
方法的精密度和准确度在0.5 ppm和5 ppm浓度下进行评估(表5)。检测的精密度通过0.5 ppm和5 ppm浓度下6次重复测定的%RSD来评估。检测浓度下的%RSD均小于2%。准确度通过与标称值的偏差百分比来评估,偏差均在10%以内。
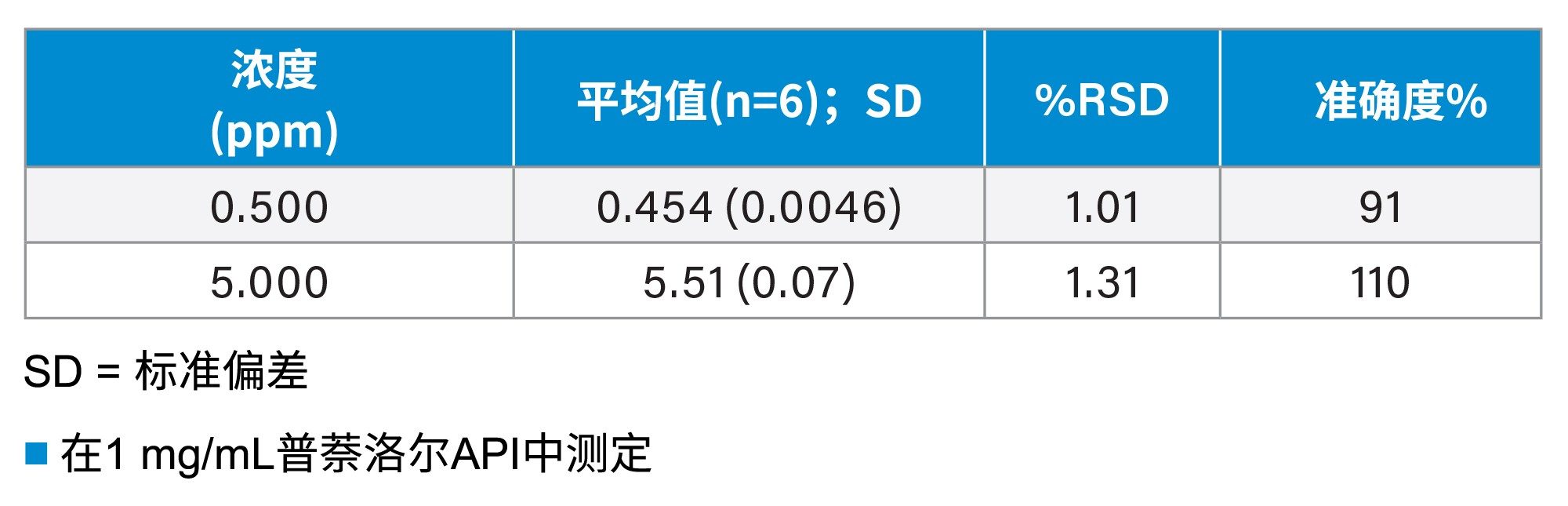 表5. 0.5 ppm和5 ppm浓度下的精密度和准确度汇总。
表5. 0.5 ppm和5 ppm浓度下的精密度和准确度汇总。
基质效应通过比较纯标准品的响应与后加标样品的校正后响应,在0.05 ppm、0.5 ppm和5 ppm浓度下进行了评估。所有测试浓度下的基质效应均< 10%。
结论
本研究开发了一种用于定量分析药物中的N-亚硝基普萘洛尔的UPLC-MS/MS方法。该方法具有很高的灵敏度,可进行高精密度的定量分析,且方法的准确性在规定的回收率范围内,符合监管指南的要求。此外,杂质N-甲酰基普萘洛尔能够与N-亚硝基普萘洛尔实现色谱分离,从而避免N-亚硝基-普萘洛尔的MRM通道(289>145和289>103)中13C同位素信号的重叠。色谱分离度对于保持分析的特异性非常重要,有助于降低误报杂质水平的风险。
参考资料
- Montesano, R.; Hall, J. Nitrosamine Metabolism and Carcinogenesis.In Mutation, Cancer, and Malformation, Chu, E. H. Y., Generoso, W. M. Eds.; Springer US, 1984; pp 447–464.
- Holzgrabe, U. Nitrosated Active Pharmaceutical Ingredients - Lessons Learned? J Pharm Sci 2023, 112 (5), 1210–1215.DOI: 10.1016/j.xphs.2023.01.021 From NLM.
- Moser, J.; Schlingemann, J.; Saal, C. N-Nitrosamines Impurities in Pharmaceuticals The Abrupt Challenges that Resulted, the Evolving Science, and the Regulatory Framework.Journal of Pharmaceutical Sciences 2023, 112 (5), 1161–1162.DOI: https://doi.org/10.1016/j.xphs.2023.01.016.
- Teasdale, A.; Moser, J.; Ford, J. G.; Creasy, J. N -Nitrosamines.In Mutagenic Impurities, Teasdale, A. Ed.; Wiley, 2021; pp 269–320.
- Bharate, S. S. Critical Analysis of Drug Product Recalls due to Nitrosamine Impurities.Journal of Medicinal Chemistry 2021, 64 (6), 2923–2936.DOI: 10.1021/acs.jmedchem.0c02120.
- Charoo, N. A.; Dharani, S.; Khan, M. A.; Rahman, Z. Nitroso Impurities in Drug Products: An Overview of Risk Assessment, Regulatory Milieu, and Control Strategy.AAPS PharmSciTech 2023, 24 (2), 60.DOI: 10.1208/s12249–023–02523–w.
- Nitrosamines EMEA-H-A5(3)-1490 - QA Art.5(3) Implementation_ QA10_revision 18_Oct23 (europa.eu) [accessed October 16th, 2023].
- Updated Information | Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs) | FDA [accessed October 16th, 2023].
- Burns, M. J.; Ponting, D. J.; Foster, R. S.; Thornton, B. P.; Romero, N. E.; Smith, G. F.; Ashworth, I. W.; Teasdale, A.; Simon, S.; Schlingemann, J. Revisiting the Landscape of Potential Small and Drug Substance Related Nitrosamines in Pharmaceuticals.Journal of Pharmaceutical Sciences.DOI: 10.1016/j.xphs.2023.10.001 (accessed October 16th, 2023).
- Pfizer Recalls Inderal-LA (propranolol hydrochloride) Capsules due to a Nitrosamine Impurity.Pfizer Recalls Inderal-LA (propranolol hydrochloride) Capsules due to a Nitrosamine Impurity - Canada.ca.[accessed October 16th, 2023].
- Li, X.; Le, Y.; Seo, J.-E.; Guo, X.; Li, Y.; Chen, S.; Mittelstaedt, R. A.; Moore, N.; Guerrero, S.; Sims, A.; et al.Revisiting the Mutagenicity and Genotoxicity of N-nitroso Propranolol in Bacterial and Human In Vitro Assays.Regulatory Toxicology and Pharmacology 2023, 141, 105410.DOI: https://doi.org/10.1016/j.yrtph.2023.105410.
720008171ZH,2024年1月