使用快速高灵敏度的LC-MS/MS生物分析方法同时定量大鼠血浆中的吉非替尼-PROTAC–3和吉非替尼,为发现DMPK研究提供支持
摘要
PROTAC分子代表了一种全新的候选药物设计方法,它能够克服许多小分子疗法所面临的耐药性问题,拓展了对传统上不可成药的蛋白质的治疗可能性。此外,与蛋白质生物治疗药物相比,PROTAC分子还具有降低生产成本、减少规模化生产难题、延长保质期、提高稳定性以及改善储存条件等优势。准确定量分析血液衍生体液中的PROTAC分子对于发现和开发DMPK方法包至关重要。本研究开发出一种快速(4 min)的LC-MS/MS生物分析方法,用于定量分析大鼠血浆中的PROTAC-3-吉非替尼和吉非替尼。测得10 μL样品中吉非替尼-PROTAC-3的检测限(LOD)为20 pg/mL,线性动态范围为20 pg/mL–1,000 ng/mL。该分析方法进行了为期三天的验证,LOD下的CV为5%。
优势
血浆中的PROTAC定量分析、LC-MS/MS、DMPK、生物分析。
简介
蛋白水解靶向嵌合体(PROTAC)是一类新型药物分子,它们通过动员泛素-蛋白酶体系统,利用细胞自身的机制,对靶蛋白实现蛋白酶体介导的降解。PROTAC主要包含三个组成部分:i)靶点结合部分,ii)连接子,以及iii)泛素E3连接酶结合部分,如图1所示。这些异双功能分子的作用机制是:靶点部分结合靶点蛋白(POI),与此同时,E3泛素连接酶与PROTAC分子的另一端结合。POI与连接酶结合后发生泛素化,然后通过细胞内的泛素-蛋白酶体系统降解,在此过程中PROTAC分子被循环利用。这些PROTAC可以被视为“较大的小分子”,因此与小分子具有许多共同属性,例如合成规模放大、生产成本、保质期、稳定性和给药途径。PROTAC通过降解消除靶蛋白的所有功能,因此提供了差异化的药理学效果,并且不需要靶点结合基团,直接抑制蛋白质功能。它们还能显著拓展“可成药”蛋白质的范围,为开发更安全的新型药物开辟了可能性1,2。
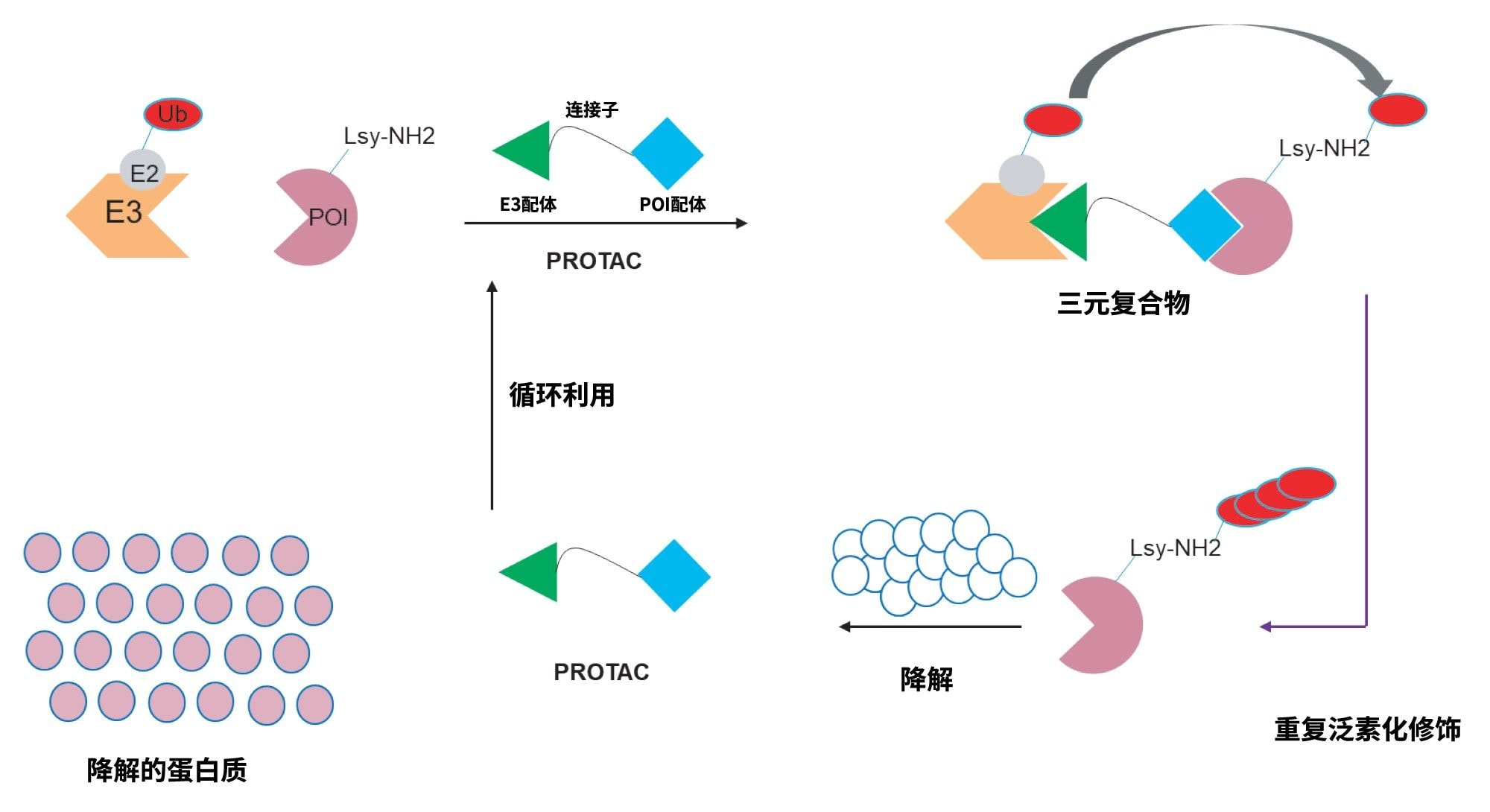 图1.PROTAC催化的蛋白质降解
图1.PROTAC催化的蛋白质降解
吉非替尼是一种酪氨酸激酶抑制剂,用于治疗非小细胞肺癌,且是一种有效的表皮生长因子受体(EGFR)抑制剂,通过阻断细胞信号传导发挥作用3。 吉非替尼在大鼠和小鼠体内的药代动力学和体内代谢特征已有研究报告,结果表明该化合物吸收良好,峰浓度出现在给药后1小时,估计的口服生物利用度为50–60%,半衰期为3.8 h(IV途径为2.6 h)3,4。 此前发表的文章已证实,PROTAC分子的半衰期较长(11–15 h),给药后48小时仍可检测到药物浓度。为了更好地测定吉非替尼-PROTAC-3的药代动力学,我们开发了一种高灵敏度生物分析方法,可同时测定大鼠血浆中的吉非替尼和吉非替尼-PROTAC-3,浓度范围为20 pg/mL~1000 ng/mL。
实验
样品描述
使用对照Wistar大鼠血浆稀释吉非替尼和吉非替尼-PROTAC-3(图2)的确证标准品,制备标准曲线,浓度范围为10 pg/mL~1,000 ng/mL。方法的开发和验证依据2018年5月FDA发布的行业生物分析方法验证指南5。 因此,确证标准品加标溶液中的有机溶剂体积保持在5% (v/v)以下。质控品(QC)通过单独称量确证标准品以类似的方式制备。在1.5 mL微量离心管中,将20 μL血浆与60 μL含有吉非替尼-d6(浓度为50 ng/mL)作为内标的乙腈混合,制备血浆样品。将溶液涡旋混合,并在-20 °C下储存1 h,再次涡旋混合,然后在4 °C下以25,000 g离心5 min。将得到的样品转移到自动进样器样品瓶中进行分析。使用1/X加权的线性回归,通过内标定量方法估算标准品和QC的反算浓度。
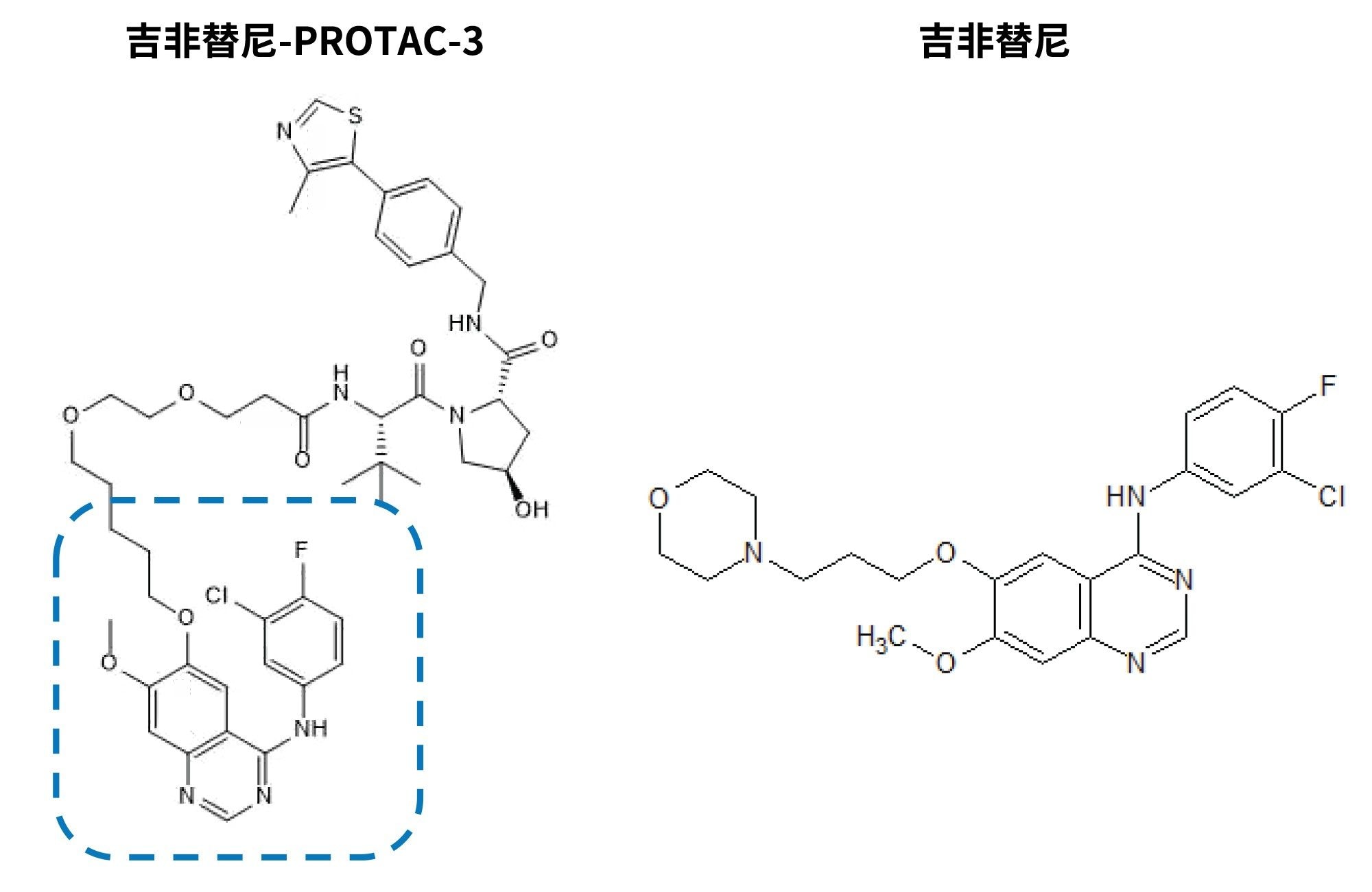 图2.PROTAC-3吉非替尼(虚线部分为吉非替尼组分)和吉非替尼
图2.PROTAC-3吉非替尼(虚线部分为吉非替尼组分)和吉非替尼
方法条件
使用ACQUITY™ UPLC™ I-Class与Xevo™ TQ-XS串联四极杆质谱仪(沃特世公司,英国威姆斯洛)的联用系统分析血浆提取物(2 μL)。本研究中采用的色谱、质谱条件和信息学方法见下表。
液相色谱条件
|
液相色谱系统: |
ACQUITY UPLC I-Class |
|
样品瓶: |
TruView LCMS认证透明螺纹颈口玻璃全回收样品瓶,P/N:186005669CV |
|
色谱柱: |
ACQUITY HSS T3™ C18 2.1 x 50 mm 1.7 µm色谱柱,P/N:186009467 |
|
柱温: |
60 °C |
|
样品温度: |
10 °C |
|
进样体积: |
2 µL |
|
流速: |
600 µL/min |
|
流动相A: |
0.1% FA (v/v)的1 mM甲酸铵水溶液 |
|
流动相B: |
0.1% FA (v/v)的ACN溶液,含1 mM甲酸铵水溶液 |
|
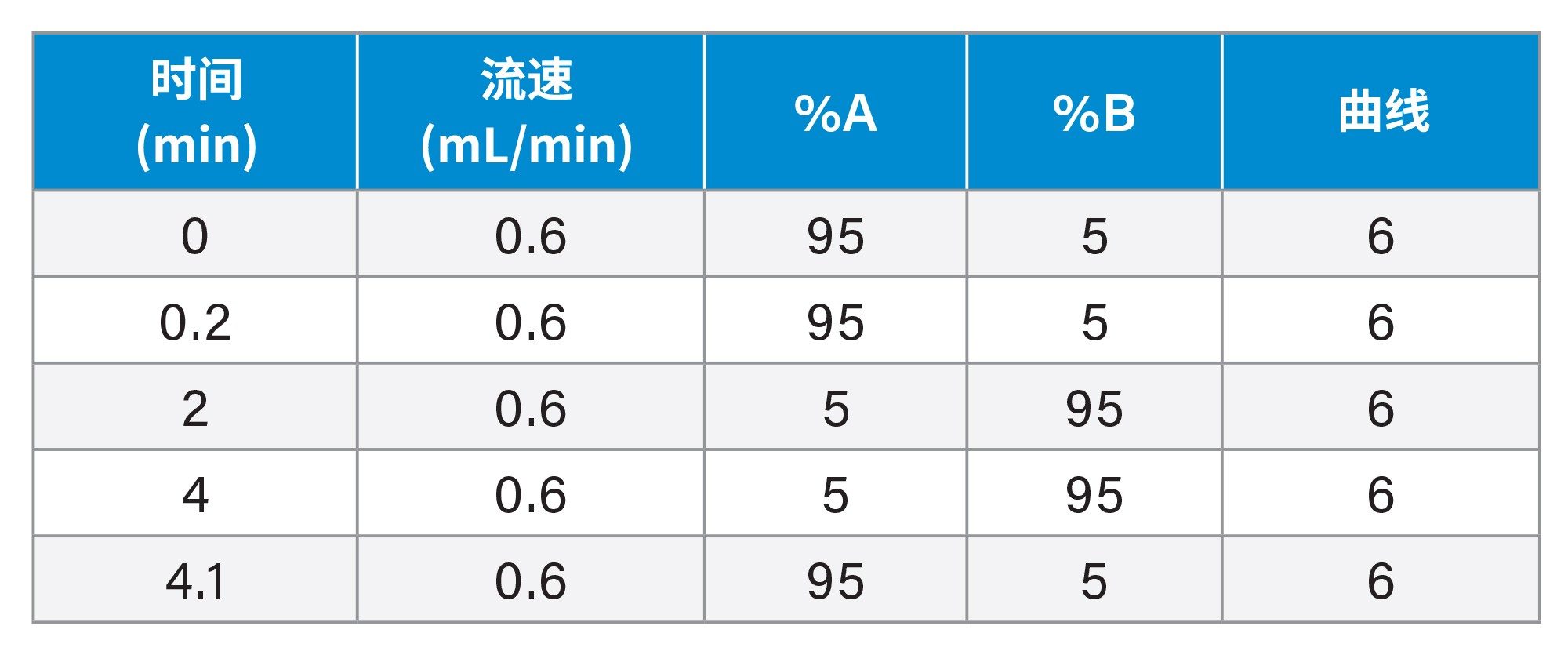
梯度: |
见下文 |
梯度表
质谱条件
|
质谱系统: |
Xevo TQ-XS三重四极杆质谱仪 |
|
电离模式: |
正离子电喷雾电离(ESI+) |
|
MRM通道: |
|
|
吉非替尼-PROTAC-3: |
m/z=934.33 > 617.34(CV=60 V,CE=34 eV) |
|
吉非替尼: |
m/z=447.25 > 128.20(CV=36 V,CE=30 eV) |
|
吉非替尼-d6: |
m/z=453.16 > 134.2(CV=50 V,CE=48 eV) |
|
毛细管电压: |
2 kV |
数据管理
|
数据采集软件: |
MassLynx™ V 4.2 |
|
数据处理软件: |
TargetLynx™ XS |
结果与讨论
本研究开发的生物分析方法可同时定量分析吉非替尼(C22H24ClFN4O3)和吉非替尼-PROTAC-3 (C47H57ClFN7O8S)。色谱分离经过优化,能够检测吉非替尼和吉非替尼-PROTAC-3的给药化合物及代谢物。使用大鼠血浆样品评估了校准范围(10 pg/mL~1,000 ng/mL)。
质谱分析
使用MassLynx Intellistart™软件评估了每种分析物的质谱MRM方法参数。各分析物溶液(100 nL/mL,50:50乙腈:水+0.1%甲酸)以10 µL/min的流速单独进样(使用质谱仪的机载流路)。在ESI+和ESI-模式下评估了母离子、子离子、锥孔电压和碰撞能量。三种分析物在ESI+模式下均显示出最强的信号响应,下面的图3显示了吉非替尼-PROTAC-3分析物的IntelliStart优化报告示例。结果表明,分析物产生质子化母离子,质荷比为m/z=934.33,并在m/z=617.34和182.06处检测到主要碎片离子。对于m/z=617.34离子,两种分析物的锥孔电压曲线相当平坦,优化为88 V,最佳碰撞能量分别确定为34和58 eV。由于通道m/z=934.17–617.34的响应更强,因此被选为吉非替尼-PROTAC-3的定量通道,使用通道m/z=447.25–128.20(CV=36 V,CE=30 eV)监测吉非替尼,使用通道m/z==453.29–134.25(CV=50 V,CE=48 eV)监测吉非替尼-d6。用于吉非替尼浓度采集的质谱采集条件采用了Molly等人先前报道的使用串联四极杆质谱仪的条件,这些条件已在上面的方法学部分列出4。
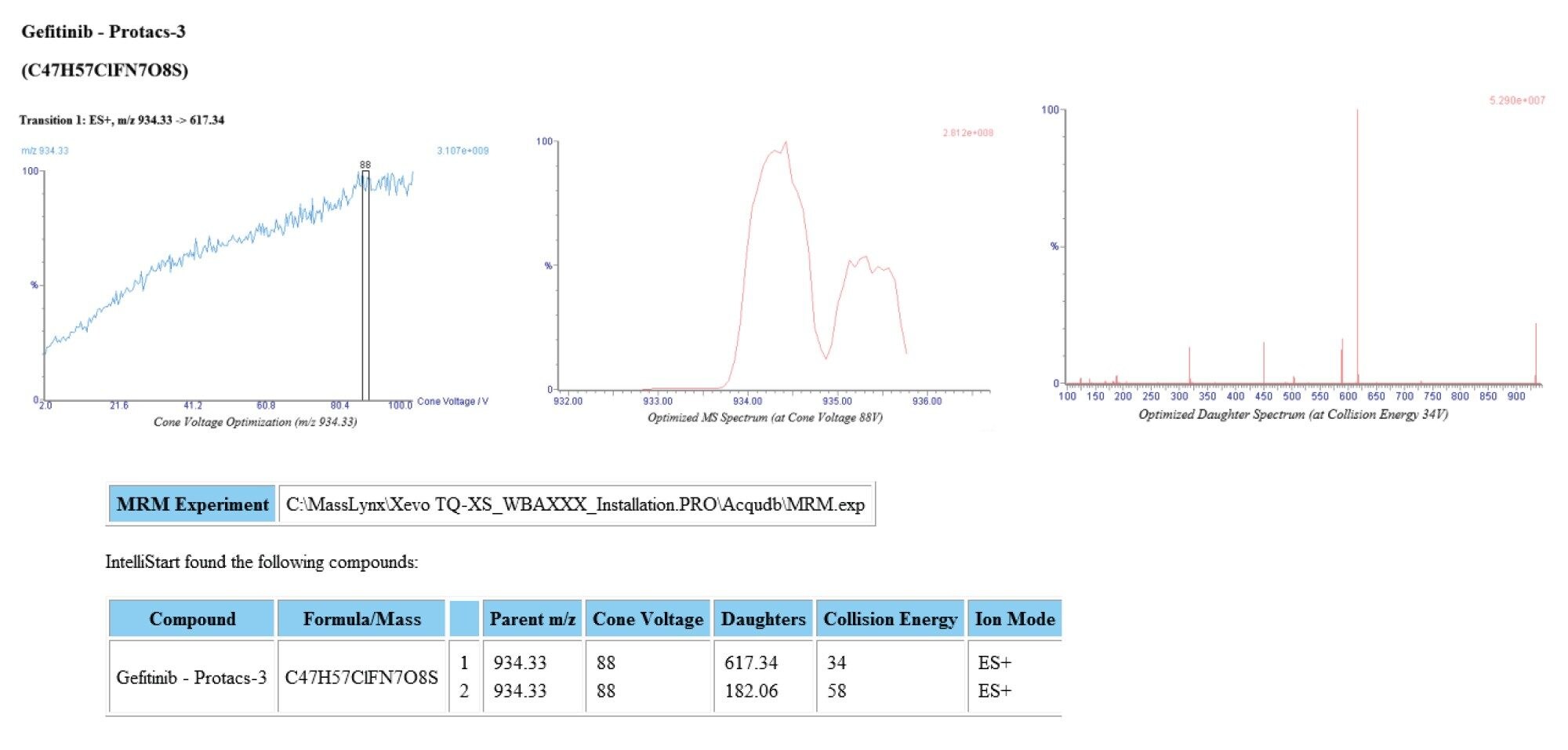 图3.吉非替尼-PROTAC-3的Intellistart正离子ESI MRM优化
图3.吉非替尼-PROTAC-3的Intellistart正离子ESI MRM优化
色谱分析
先前针对吉非替尼体内代谢的研究确定了几种极性代谢物,为防止这些代谢物与吉非替尼共洗脱,需要使用低有机相组成起始的反相梯度3,4。 而吉非替尼-PROTAC-3需要使用高有机相浓度的流动相才能实现洗脱。因此,为了分离吉非替尼、吉非替尼-PROTAC-3以及任何潜在生物转化,我们对色谱分析进行了优化。最终条件采用ACQUITY UPLC HSS T3色谱柱,柱温60 °C,梯度为:2 min内5–95%乙腈-水相,95%乙腈保持2 min,然后在4.1 min返回初始条件,流速为600 μL/min。在这些条件下,吉非替尼和吉非替尼-PROTAC-3洗脱的保留时间tR分别为1.40 min和1.86 min。
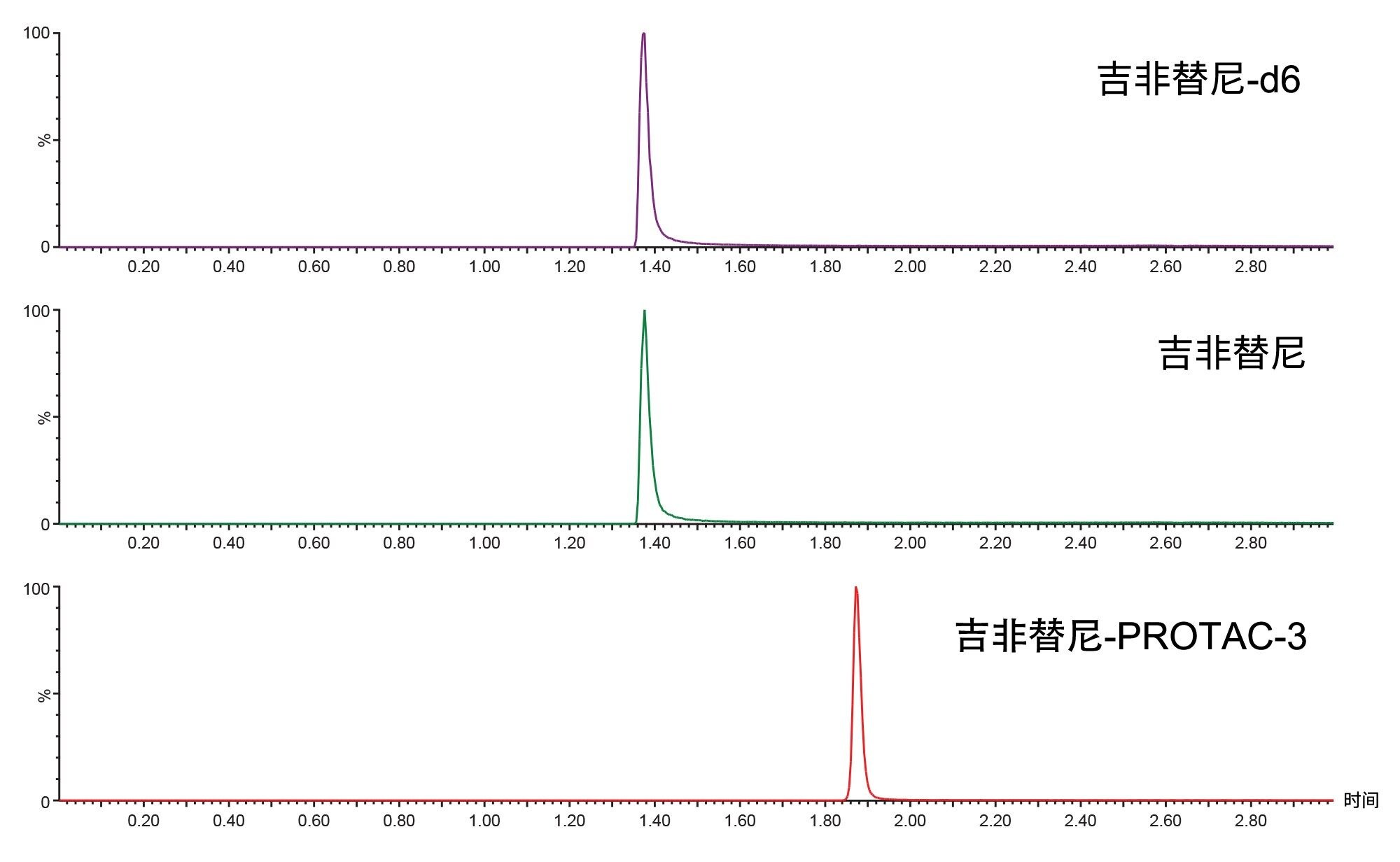 图4.使用2.1 x 50 mm ACQUITY HSS T3 1.7 µm C18色谱柱在正离子模式下对吉非替尼-PROTAC-3、吉非替尼-d6和吉非替尼进行LC-MS/MS分析,柱温维持在60 °C,采用5–95%水–乙腈(含1 mM甲酸铵和0.1%甲酸)的梯度洗脱,梯度时间为2分钟,并保持2分钟。
图4.使用2.1 x 50 mm ACQUITY HSS T3 1.7 µm C18色谱柱在正离子模式下对吉非替尼-PROTAC-3、吉非替尼-d6和吉非替尼进行LC-MS/MS分析,柱温维持在60 °C,采用5–95%水–乙腈(含1 mM甲酸铵和0.1%甲酸)的梯度洗脱,梯度时间为2分钟,并保持2分钟。
样品前处理
如前文所述,使用1:3的样品(20 μL)与有机溶剂比率,通过蛋白沉淀法制备血浆样品。
校准范围
在10 pg/mL~1,000 ng/mL的范围内评估了吉非替尼-PROTAC-3方法的检测下限(LLoD)和可用线性动态范围。大鼠血浆中的检测限为20 pg/mL(柱上进样量2.5 fg),代表性色谱图见图5。使用内标定量方法和1/x加权,确定标准曲线的线性范围约为5个数量级(20 pg/mL–1,000 ng/mL)。代表性标准曲线见图6,相关系数r2=0.998,负值截距为-152.165。
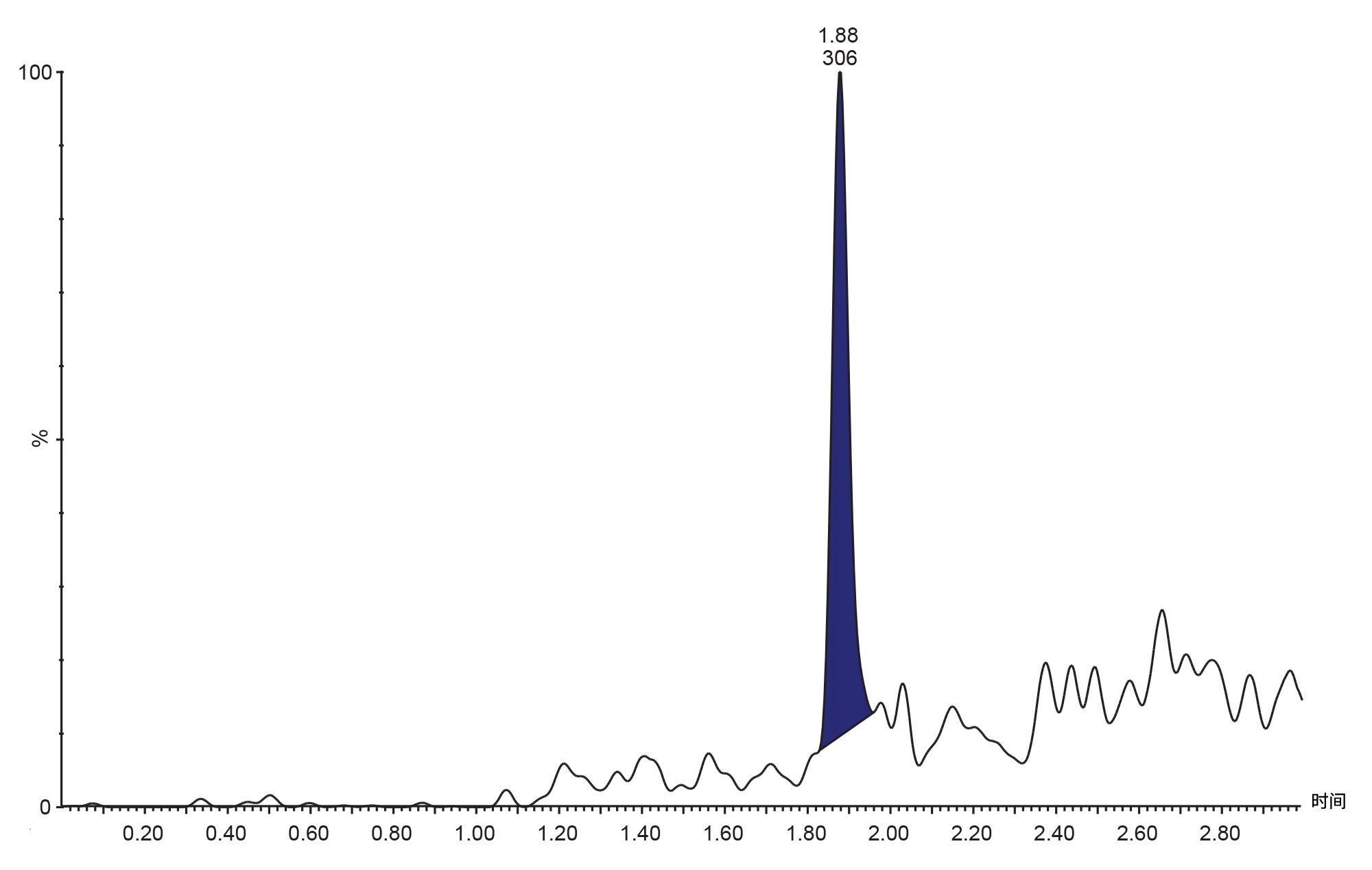 图5.使用2.1 x 50 mm ACQUITY HSS T3 1.7 µm C18色谱柱在正离子模式下对20 pg/mL吉非替尼-PROTAC-3标准品进行LC-MS/MS分析,柱温维持在60 °C,采用5–95%水–乙腈(含1 mM甲酸铵和0.1%甲酸)的梯度洗脱,梯度时间为2分钟,并保持2分钟。
图5.使用2.1 x 50 mm ACQUITY HSS T3 1.7 µm C18色谱柱在正离子模式下对20 pg/mL吉非替尼-PROTAC-3标准品进行LC-MS/MS分析,柱温维持在60 °C,采用5–95%水–乙腈(含1 mM甲酸铵和0.1%甲酸)的梯度洗脱,梯度时间为2分钟,并保持2分钟。
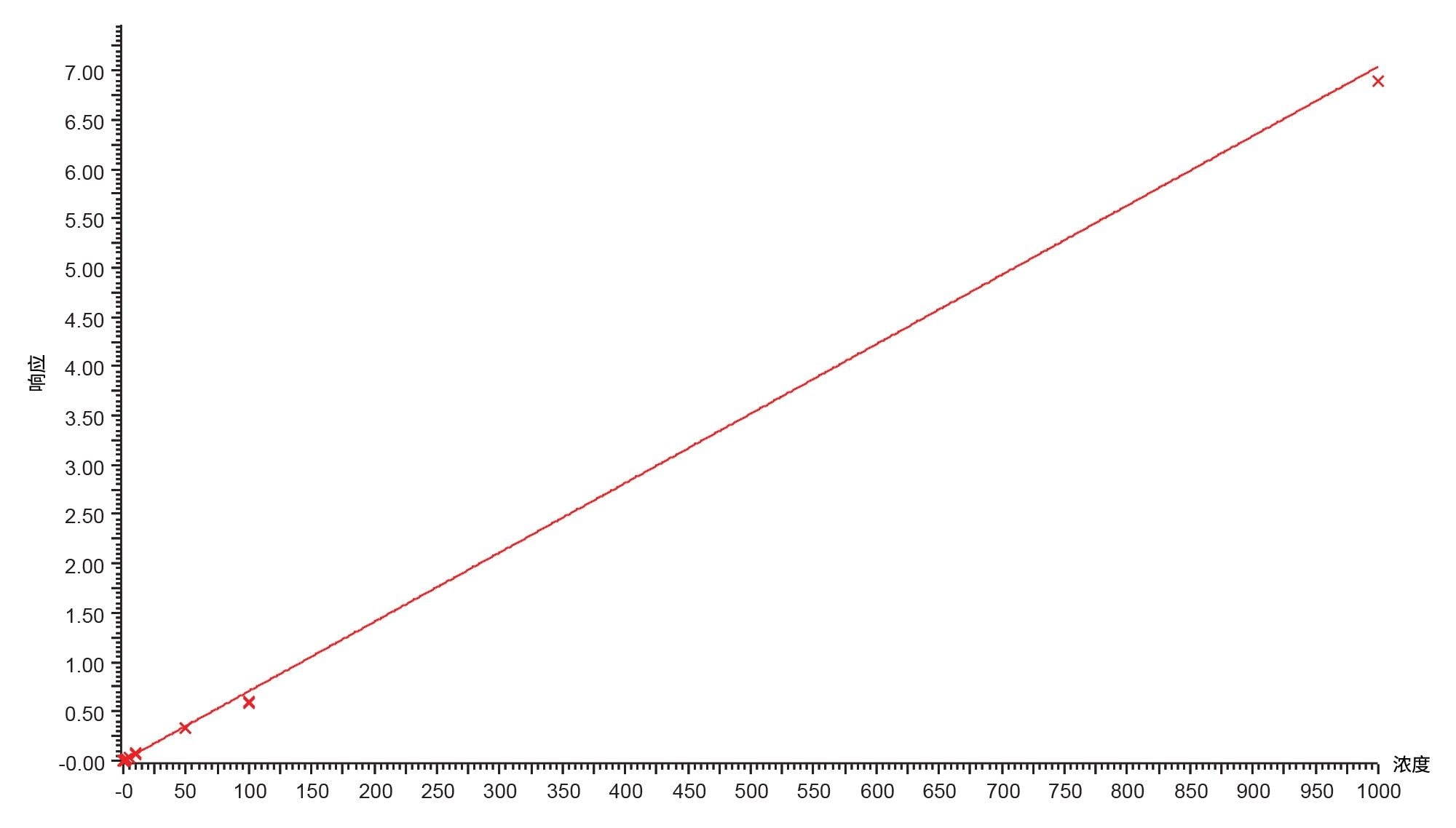 图6.吉非替尼-PROTAC-3的标准曲线,范围为20 pg/mL~1000 ng/mL
图6.吉非替尼-PROTAC-3的标准曲线,范围为20 pg/mL~1000 ng/mL
测得吉非替尼的检测限为0.05 ng/mL,相关系数为0.997,大鼠血浆提取物中吉非替尼的典型标准曲线和LOQ标准品如图7所示。
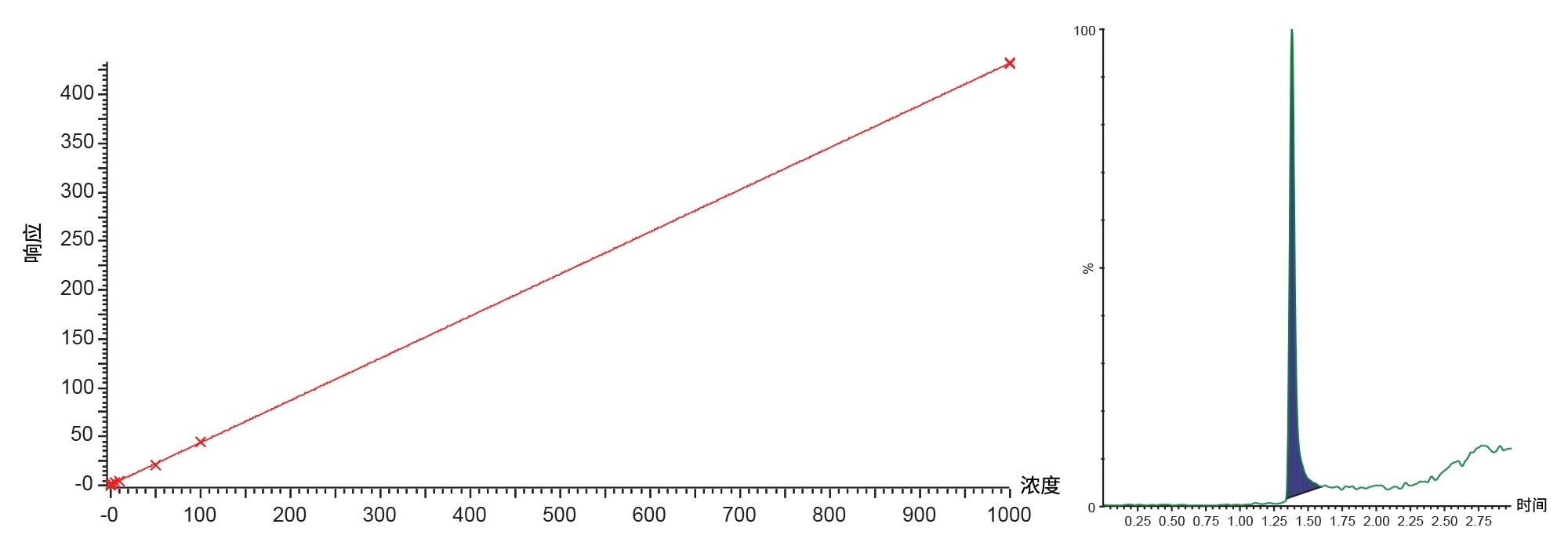 图7.吉非替尼的标准曲线(范围为0.5~1000 ng/mL)和LOQ标准品(0.5 ng/mL)
图7.吉非替尼的标准曲线(范围为0.5~1000 ng/mL)和LOQ标准品(0.5 ng/mL)
为期三天的验证研究
之前的研究表明,小鼠和大鼠PO给药后吉非替尼吸收良好,在给药后1小时观察到血浆峰浓度,以50 mg/kg的剂量向小鼠给药时,峰浓度值为7 µg/mL4。 血浆浓度迅速下降,半衰期为3.8 h,给药后24 h几乎检测不到药物。因此,在剂量水平通常为1-10 mg/kg的典型临床前安全性评估研究中,选择0.5-1000 ng/mL的分析浓度范围,对吉非替尼和吉非替尼-PROTAC-3的生物分析方法进行了为期三天的验证。在三天内按以下顺序分析三批样品:溶剂空白;空白;标准曲线(0、0.5、1、2、5、10、20、50、100、500和1000 ng/mL);基质空白;QC(3、75、800 ng/mL)样品;空白;提取的血浆样品;QC(3、75和800 ng/mL);空白;标准曲线(0、0.5、1、2、5、10、20、50、100、500和1000 ng/mL);空白。为期三天的验证结果表明,两种化合物均表现出优异的重现性和准确度,如表1和表2所示。
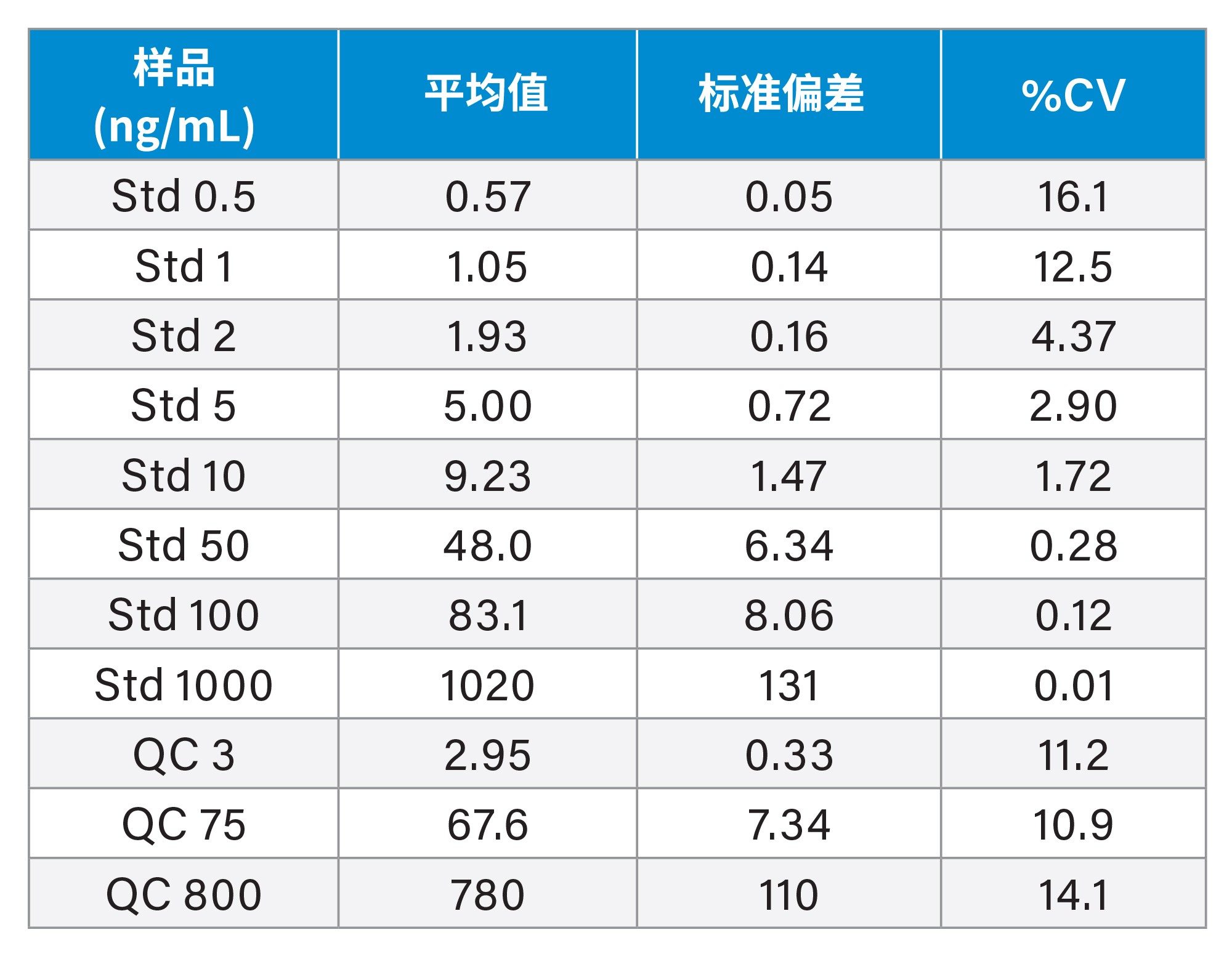 表1.在为期三天的验证中,吉非替尼-PROTAC-3的定量分析结果总结(每种标准品和QC共进行6次分析)
表1.在为期三天的验证中,吉非替尼-PROTAC-3的定量分析结果总结(每种标准品和QC共进行6次分析)
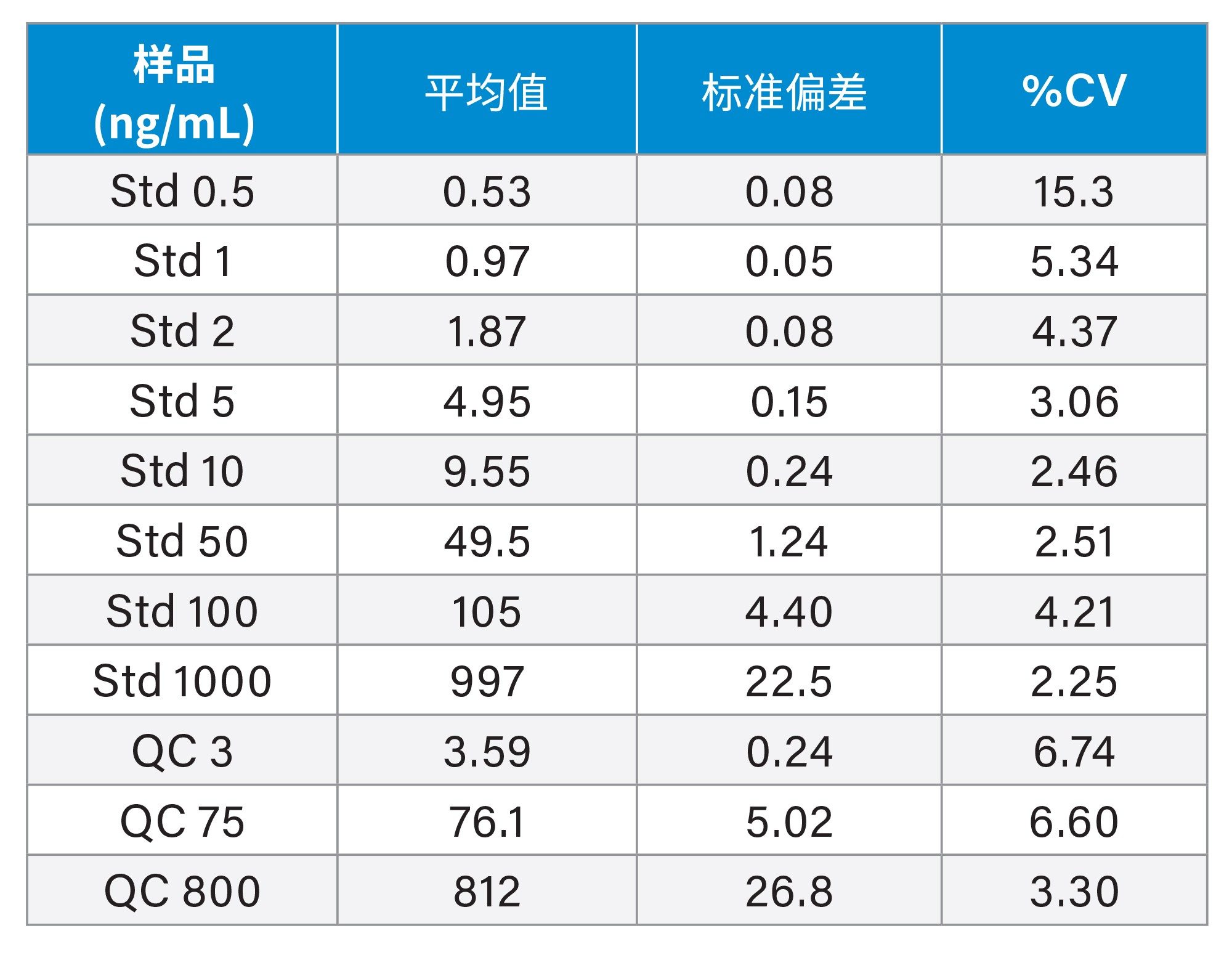 表2.在为期三天的验证中,吉非替尼的定量分析结果总结(每种标准品和QC共进行6次分析)
表2.在为期三天的验证中,吉非替尼的定量分析结果总结(每种标准品和QC共进行6次分析)
通过分析QC样品(3、75和800 ng/mL),测定吉非替尼和吉非替尼-PROTAC-3的冻融稳定性。样品制备完成后,在-20 °C下进行三次冻融循环,然后直接分析。结果表明,吉非替尼和吉非替尼-PROTAC-3的实测浓度准确度%范围分别为102.9%-108.1%和69.7%-109.1%。
结论
蛋白水解靶向嵌合体(PROTAC)通过与泛素E3连接酶结合的蛋白质靶向配体,实现蛋白酶体介导的靶蛋白降解,其分子量范围为700–1000 Da。在药物发现和开发过程中,需要确定PROTAC分子的DMPK特性,其研究方法与所有小分子和生物制剂类似;因此需要一种高灵敏度的生物分析方法。本研究使用沃特世串联四极杆质谱仪开发并验证了一种反相梯度LC-MS/MS生物分析方法,用于定量分析大鼠血浆中的吉非替尼-PROTAC-3。检测限为20 pg/mL(柱上进样量2.5 fg),动态范围为0.02~1,000 ng/mL。在1~1,000 ng/mL浓度范围内对该方法进行了为期三天的验证。
参考资料
- Bhole RP, Kute PR, Chikhale RV, Bonde CG, Pant A, Gurav SS.PROTACs: A Comprehensive Review of Protein Degradation Strategies in Disease Therapy. Bioorg Chem. 2023;139:106720.doi: 10.1016/j.bioorg.2023.106720.
- Békés M, Langley DR, Crews CM.PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat Rev Drug Discov. 2022;21(3):181–200.doi: 10.1038/s41573-021-00371-6.
- McKillop D, Hutchison M, Partridge EA, Bushby N, Cooper CM, Clarkson-Jones JA, Herron W, Swaisland HC. Metabolic Disposition of Gefitinib, an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, in Rat, Dog and Man.Xenobiotica.2004;34(10):917–34.doi: 10.1080/00498250400009171.
- Molloy B, Mullin L, King A, Gethings LA, Plumb RS, Wilson ID.The Pharmacometabodynamics of Gefitinib After Intravenous Administration to Mice: A Preliminary UPLC-IM-MS Study.Metabolites.2021 11;11(6):379.doi: 10.3390/metabo11060379.
- https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf
720008229ZH,2024年1月