在临床研究中使用UPLC-MS/MS分析尿液中的游离变肾上腺素
仅供研究使用,不适用于诊断。
摘要
本应用纪要展示了Xevo TQ-XS IVD质谱仪在临床研究中用于分析尿液中的游离变肾上腺时表现出的优异灵敏度和定量性能。
优势
- 使用反相色谱法测定尿液中的游离变肾上腺素
- 无需进行酸水解或酶解偶联步骤,而这些步骤在测定游离形式和结合形式的变肾上腺素中往往是必不可少的
- 研究人员认为,尿液中的游离变肾上腺素相比过去使用的分离的变肾上腺素具有更优异的临床性能
简介
变肾上腺素和去甲变肾上腺素是儿茶酚胺的无活性代谢物。主要的儿茶酚胺有多巴胺、肾上腺素和去甲肾上腺素,人体会在身体或者情感遭受压力时释放这些物质。它们有助于在大脑中传递神经脉冲,增加葡萄糖和脂肪酸的释放以获取能量,并扩张肺部的细小气道。去甲肾上腺素还会收缩血管,导致血压升高,而肾上腺素会增加心率并促进新陈代谢。去甲肾上腺素分解为去甲变肾上腺素,肾上腺素则分解为变肾上腺素,然后经尿液排泄。
这些代谢物通常以未结合形式(即游离形式)或与硫酸盐结合的形式存在于尿液中,数量波动不定,在身体受到压力期间和之后不久会显著增加。然而,罕见的嗜铬细胞瘤和其他神经内分泌肿瘤会产生大量儿茶酚胺,导致血液和尿液中这些激素及其代谢物的浓度大幅增加。过去,在检测嗜铬细胞瘤时,会同时对结合形式和游离形式(通常称为分离的变肾上腺素)进行定量。最近有证据表明,尿液中的游离变肾上腺素对于评估肿瘤细胞产生的变肾上腺素是一种更具特异性的标志物。测定尿样中的游离变肾上腺素时,无需进行酸水解或酶解偶联步骤,而测定尿液中分离的变肾上腺素时,这些步骤是必须的。
临床研究人员在测定尿液中的变肾上腺素时,通常对浓度升高的情况很感兴趣。但是,这些化合物由于极性较大,利用反相LC-MS/MS进行分析是一项巨大挑战。因此,许多研究实验室仍采用离子对试剂与电化学检测(ECD)分析这类物质。虽然反相LC-MS/MS已得到成功应用,但由于尿液基质组分发生离子抑制、保留作用不足和去甲变肾上腺素与肾上腺素未完全分离,致使挑战仍然存在。HILIC方法已得到成功应用,但实施起来可能充满挑战。
本文介绍了一种反相UPLC-MS/MS临床研究方法,采用混合模式SPE程序,使用Waters ACQUITY™ UPLC™ HSS™ PFP色谱柱进行反相色谱分离,并使用Xevo™ TQ-XS IVD质谱仪进行检测。该方法快速、分析灵敏度高、线性和精密度优异,基质效应非常小,适用于临床研究。
 图1.ACQUITY™ UPLC™ I-Class FTN IVD系统和Xevo TQ-XS IVD质谱仪
图1.ACQUITY™ UPLC™ I-Class FTN IVD系统和Xevo TQ-XS IVD质谱仪
实验
样品前处理和UPLC-MS/MS分析
市售标准曲线样品(6PLUS1多水平尿液标准曲线样品套装MassChrom,包括尿液中的儿茶酚胺、游离变肾上腺素、血清素)购自Chromsystems®(Gräfelfing,德国),按照制造商的说明制备。变肾上腺素的标准曲线范围为14~11,941 nmol/L,去甲变肾上腺素为27~11,031 nmol/L,3-甲氧酪胺为43~7,385 nmol/L。QC样品是ChromSystems的MassCheck儿茶酚胺、游离变肾上腺素、血清素,尿液控制水平I和II,其中变肾上腺素的浓度分别为414 nmol/L和3,427 nmol/L,去甲变肾上腺素的浓度分别为459 nmol/L和3,303 nmol/L,3-甲氧酪胺的浓度分别为399 nmol/L和1,943 nmol/L。水、乙腈和甲酸均购自Merck(英国吉林汉姆)。
变肾上腺素数据除以5.06、去甲变肾上腺素数据除以5.46、3-甲氧酪胺数据除以5.98,可将SI单位(nmol/L)转换为传统的质量单位(ng/mL)。
用110 μL含有70 ng/mL内标的水溶液对未酸化的尿液样品(110 μL )进行预处理。将2 mg 30 µm的Oasis™ WCX SPE µElution板(P/N:186002499)依次用200 μL含1%甲酸的85%乙腈和200 μL水进行活化,每步操作完成后均在正压下干燥。向每个孔中加入175 μL样品/内标混合物,并在正压条件下通过样品板。依次用200 μL水和200 μL乙腈处理每个孔,每步操作完成后同样利用正压干燥每个孔。用60 μL 85%乙腈+1%甲酸的流动相将目标分析物从吸附床上洗脱到96孔样品收集板(P/N:186002482)中。将样品挥干并复溶于500 μL水中。
随后将样品(10 µL)进样至ACQUITY UPLC I-Class FTN系统和Xevo TQ-XS IVD质谱仪中,使用水/乙腈梯度,均含0.1%甲酸,将ACQUITY UPLC HSS PFP, 2.1 x 100 mm, 1.8 µm色谱柱(P/N:186005967)加热至35 °C,如表1所示。
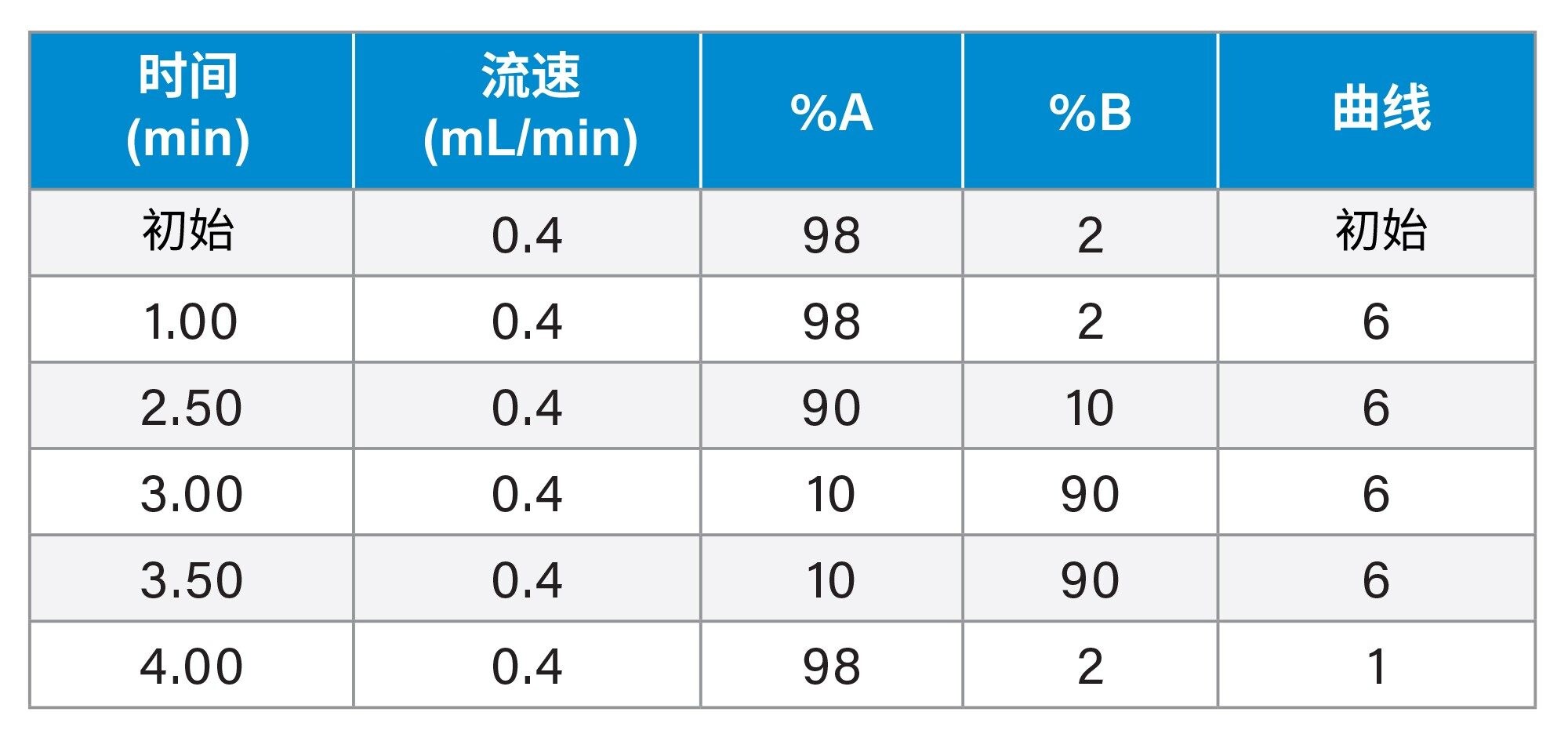 表1.分离变肾上腺素、去甲变肾上腺素和3-MT的梯度表,总运行时间为5.0 min。
表1.分离变肾上腺素、去甲变肾上腺素和3-MT的梯度表,总运行时间为5.0 min。
表2列出了检测每种分析物采用的多重反应监测(MRM)通道、定量离子(quan)和定性离子(qual),实验采用电喷雾正离子模式,毛细管电压为0.5 kV,MS1和MS2分辨率设置为0.7 FWHM。
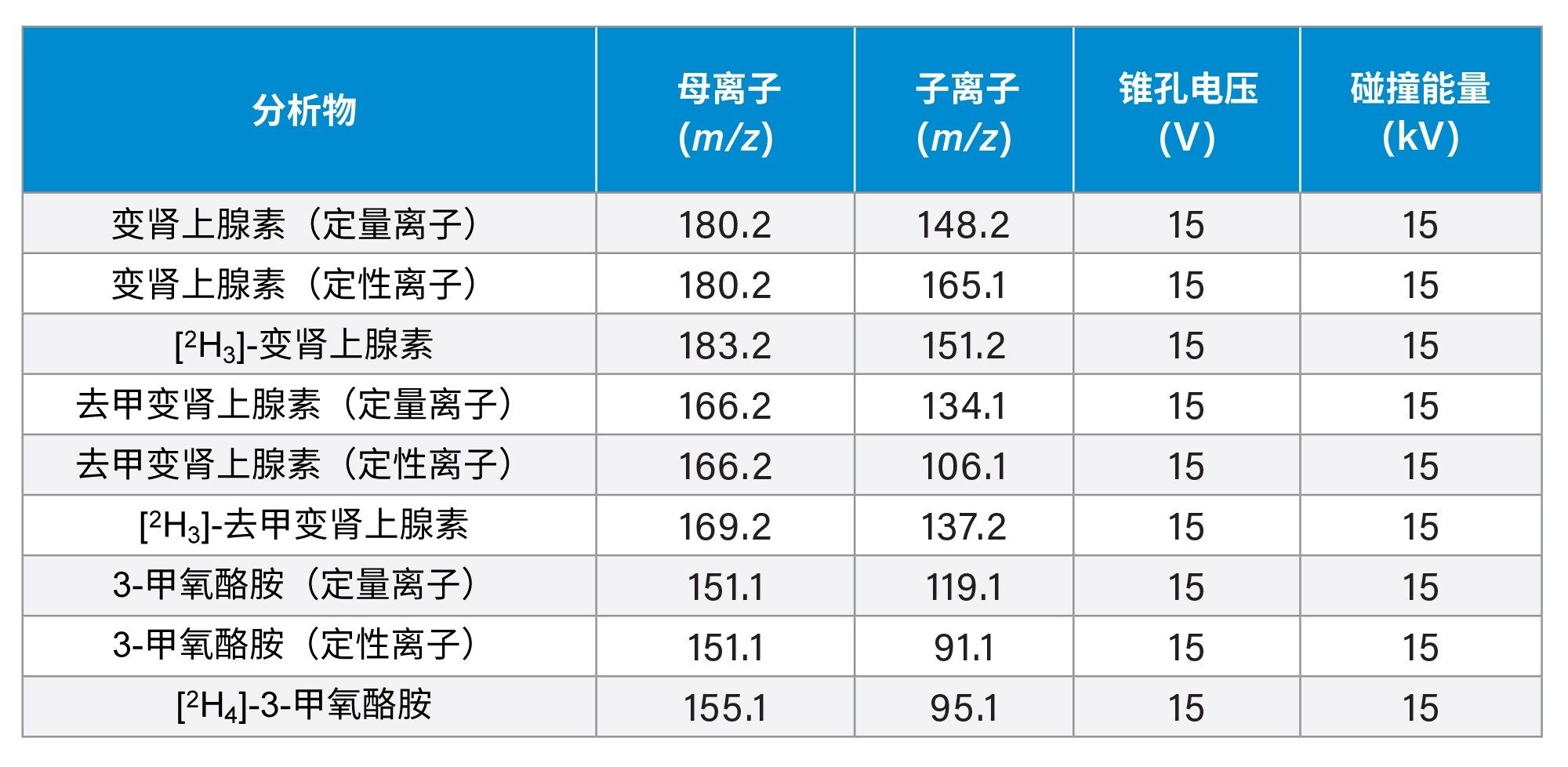 表2.变肾上腺素、去甲变肾上腺素和3-MT定量离子、定性离子及其内标的MRM参数
表2.变肾上腺素、去甲变肾上腺素和3-MT定量离子、定性离子及其内标的MRM参数
结果与讨论
变肾上腺素、去甲变肾上腺素和3-甲氧酪胺实现了色谱分离,进样间的间隔时间约为5.5 min。典型色谱图如图2所示。
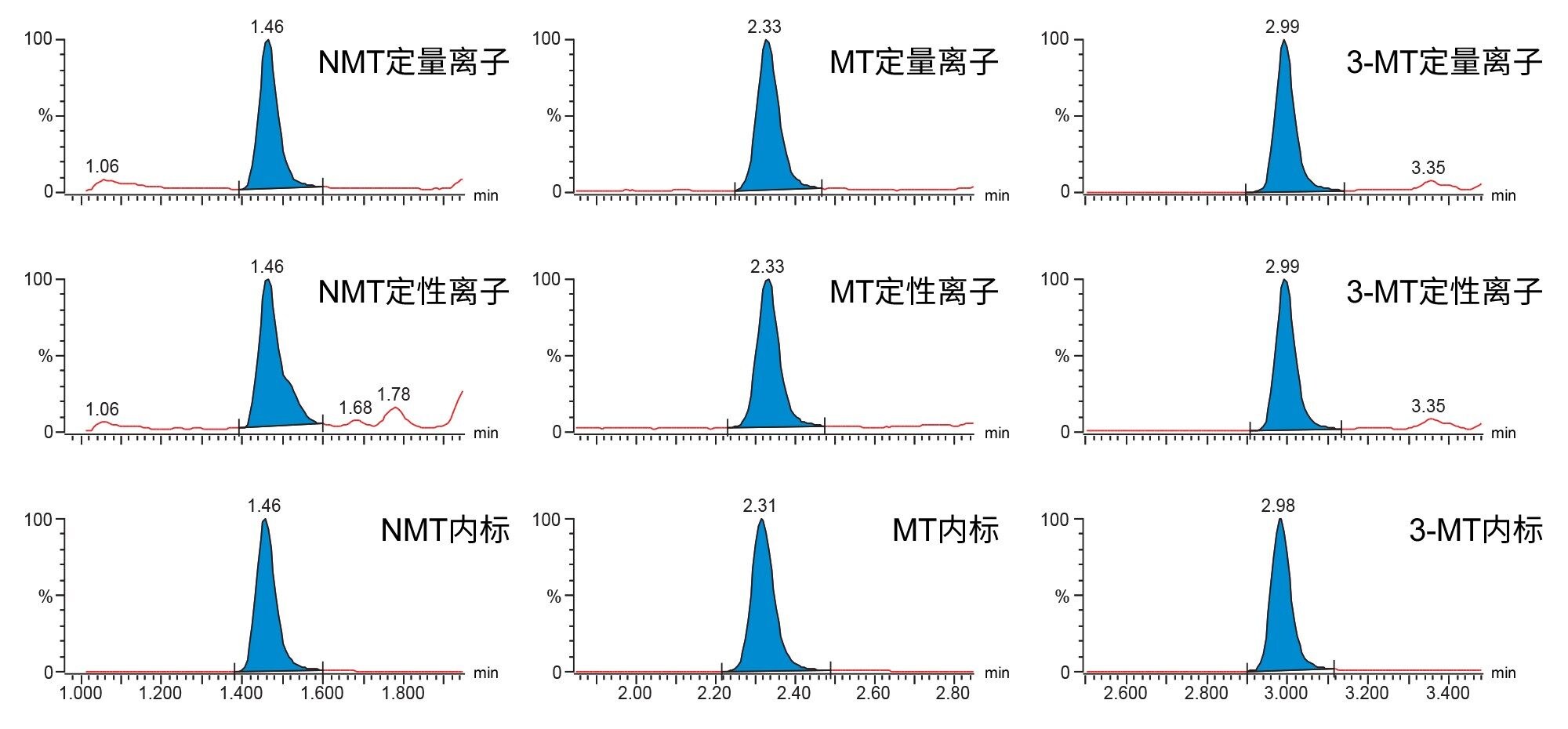 图2.标准曲线样品1中去甲变肾上腺素(NMT)、变肾上腺素(MT)和3-甲氧酪胺(3-MT)的典型色谱图(浓度分别为23.0 nmol/L、14.9 nmol/L和37.1 nmol/L)
图2.标准曲线样品1中去甲变肾上腺素(NMT)、变肾上腺素(MT)和3-甲氧酪胺(3-MT)的典型色谱图(浓度分别为23.0 nmol/L、14.9 nmol/L和37.1 nmol/L)
标准曲线在前文所述的标曲范围内呈线性,相关系数>0.99,八个数据点处变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的结果偏差(%)都在±15%以内(标曲样品1为±20%)。
根据分析浓度范围向五个单独的尿液样品中添加已知浓度的分析物标准品,测定回收率,计算公式为:(加标结果-基线结果)/添加的标准品。变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的回收率%分别为103% (88–111%)、109% (90–117%)和103% (96–111%)。
在相同条件下萃取并分析浓度由低到高的五个不同样品,每个浓度制备八个重复样,评估批次内精密度。在所有浓度水平下,所有分析物的批次内精密度均为CV≤10.7%,结果汇总于表3中。
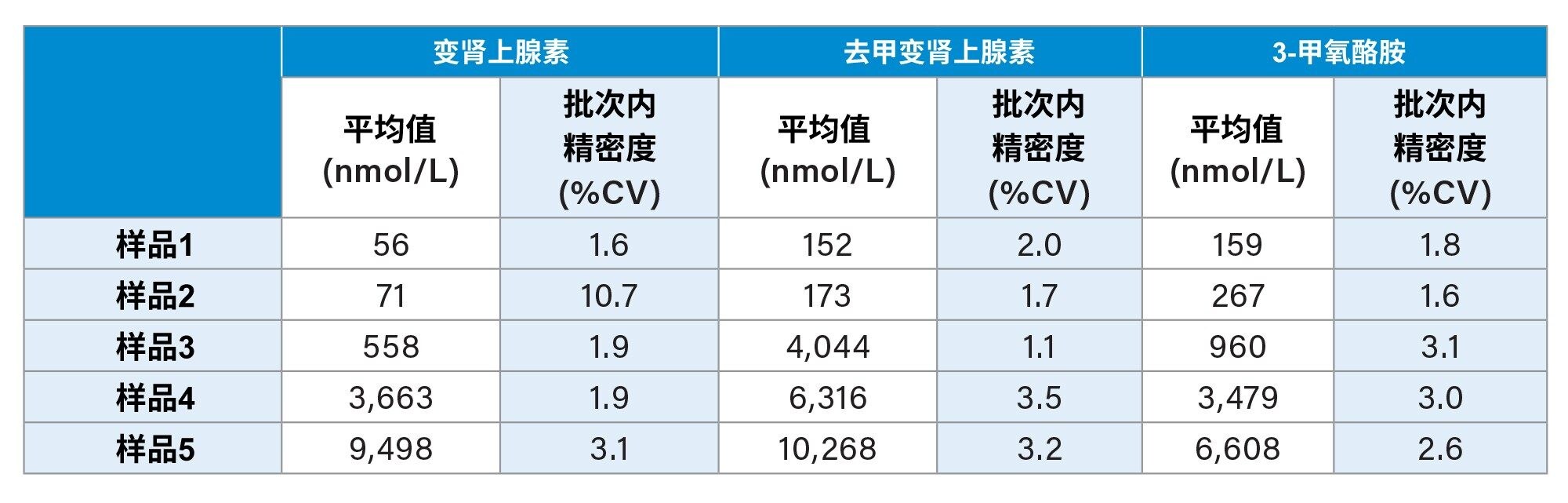 表3.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的批次内精密度性能汇总
表3.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的批次内精密度性能汇总
在五个不同的条件下萃取并分析浓度由低到高的五个不同样品,每个浓度一个重复样,评估批次间精密度。在所有浓度水平下,所有分析物的批次间精密度均为CV≤4.0%,结果汇总于表4中。
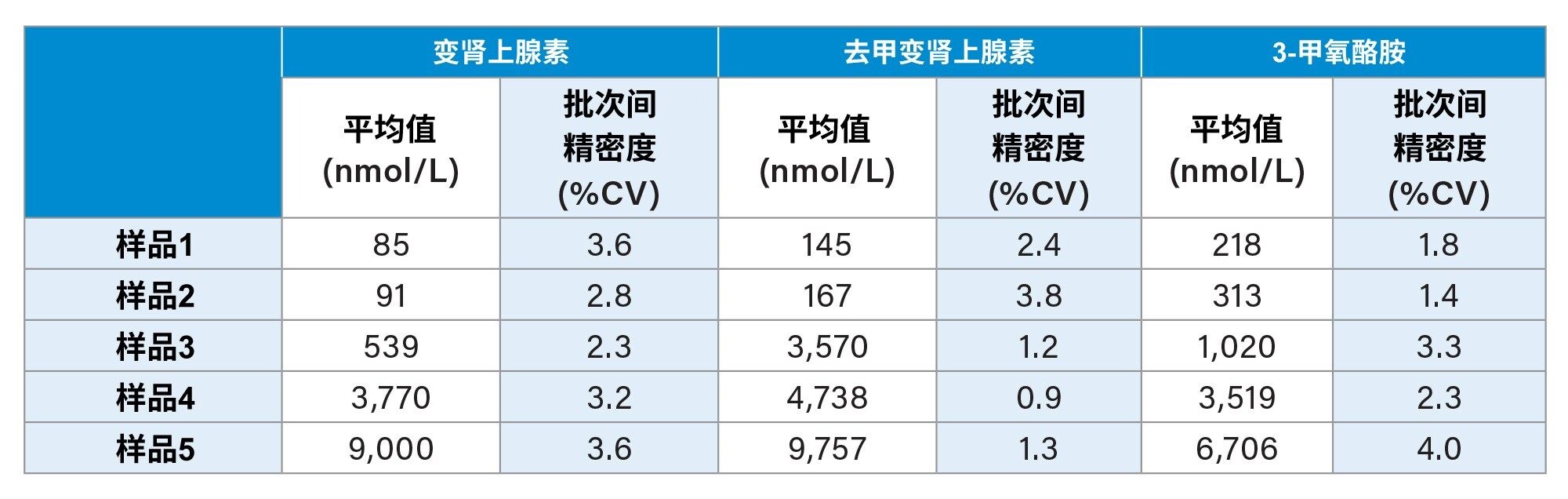 表4.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的批次间精密度性能汇总
表4.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的批次间精密度性能汇总
在比较保护性空白样品与高浓度加标样品的峰面积时,几乎没有观察到变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的残留现象。还进行了稀释测试,结果表明,除了3-甲氧酪胺外,其他样品可以稀释到1:3的比例,结果偏差在±15%以内。而3-甲氧酪胺只能稀释到1:1的比例,才能保证结果的偏差在±15%以内。此外,在4 °C下萃取后,样品可在仪器上稳定存放长达48 h。
我们使用来自六个供体的尿液样品研究了基质效应。单独定量内源性干扰物的峰面积,使用平均峰面积调整低浓度、中等浓度和高浓度萃取后加标样品的结果,以便与溶剂加标样品的结果进行比较。虽然通过峰面积可以观察到一定的基质抑制和基质增强效应,但内标可以补偿这些变化(表5)。
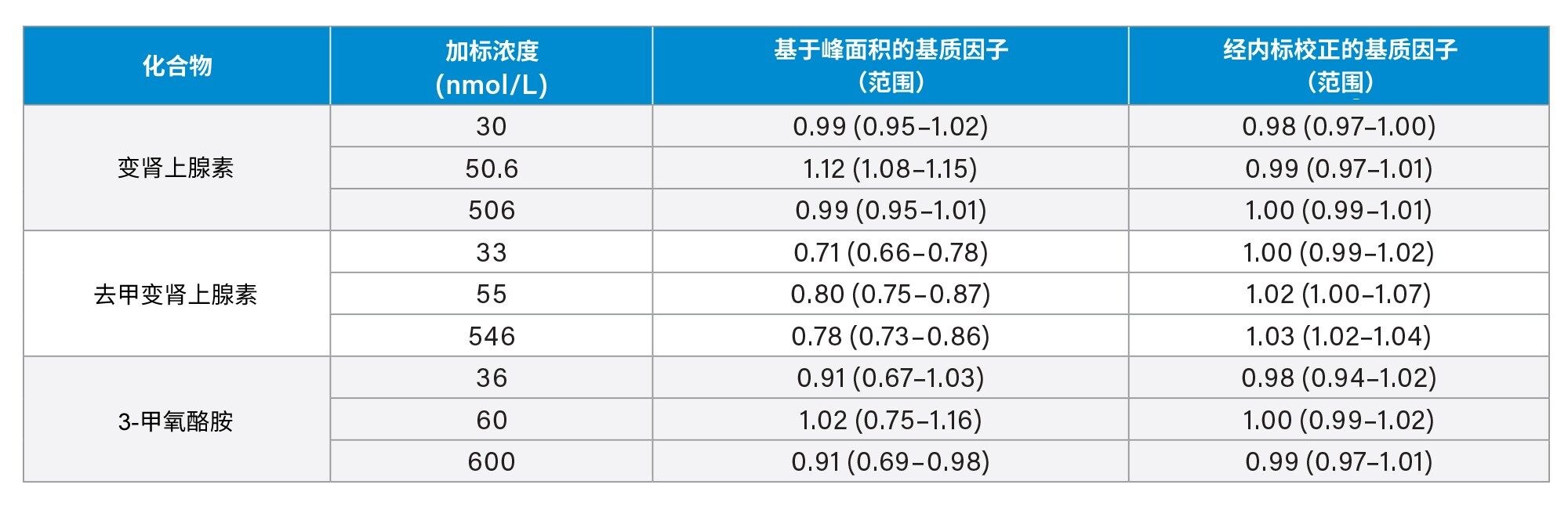 表5.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的基质因子汇总
表5.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的基质因子汇总
通过分析RCPA质量保证计划样品评估准确度,60个变肾上腺素样品、34个去甲变肾上腺素样品和30个3-甲氧酪胺样品。比较结果见表6和图3~5,与EQA计划结果表现出优异的一致性,偏差极小。
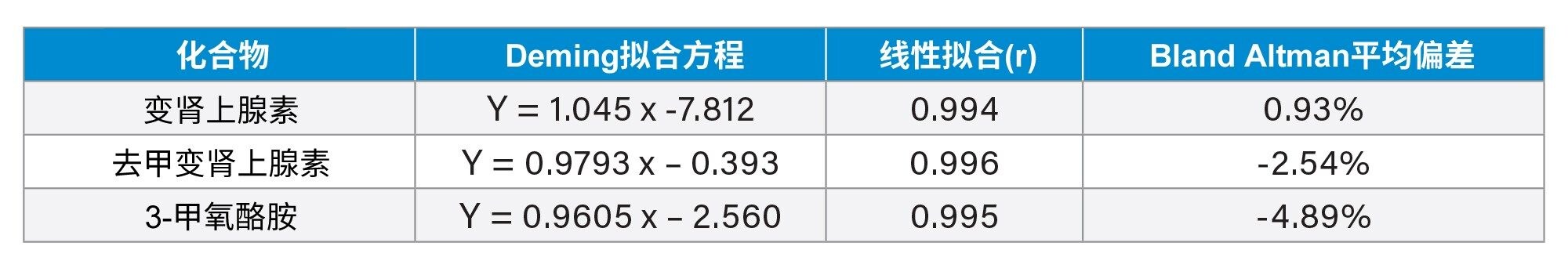 表6.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的准确度汇总
表6.变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的准确度汇总
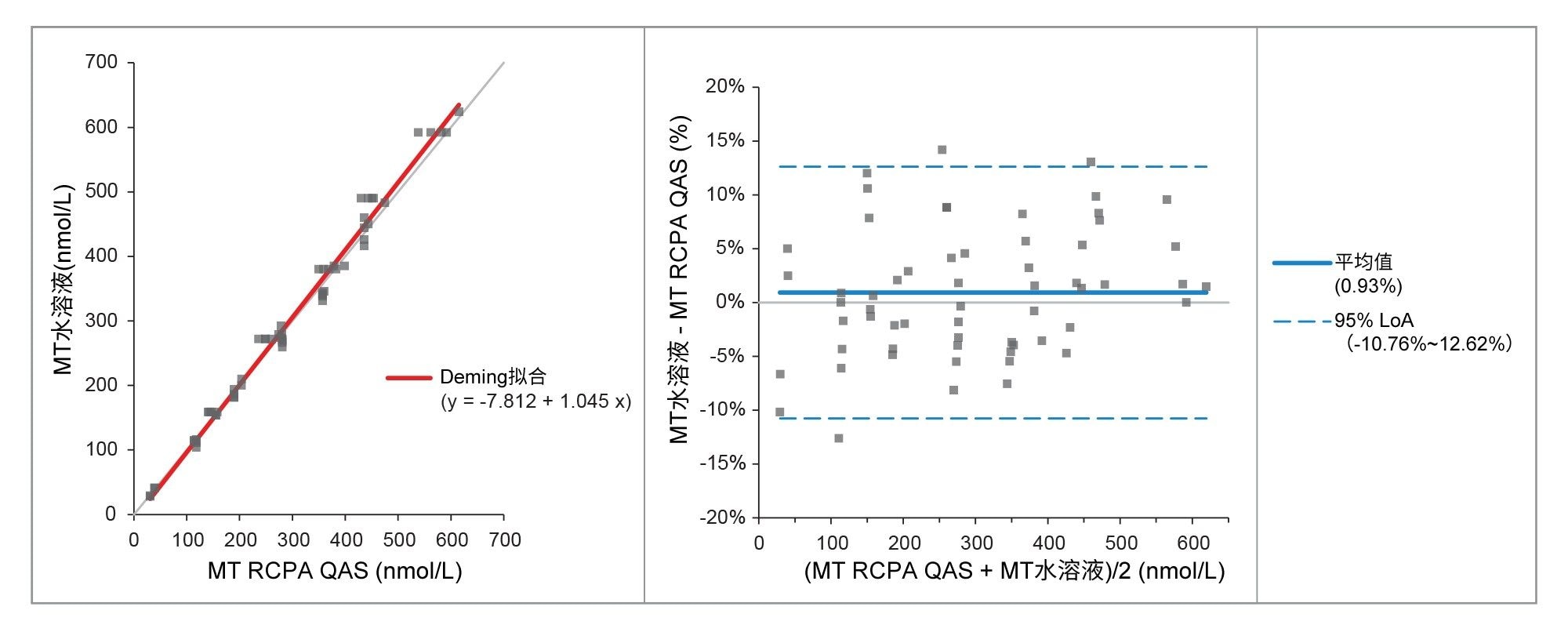 图3.比较实测值与RCPA QAS值得出的变肾上腺素Deming拟合图和Bland Altman图
图3.比较实测值与RCPA QAS值得出的变肾上腺素Deming拟合图和Bland Altman图
 图4.比较实测值与RCPA QAS值得出的去甲变肾上腺素Deming拟合图和Bland Altman图
图4.比较实测值与RCPA QAS值得出的去甲变肾上腺素Deming拟合图和Bland Altman图
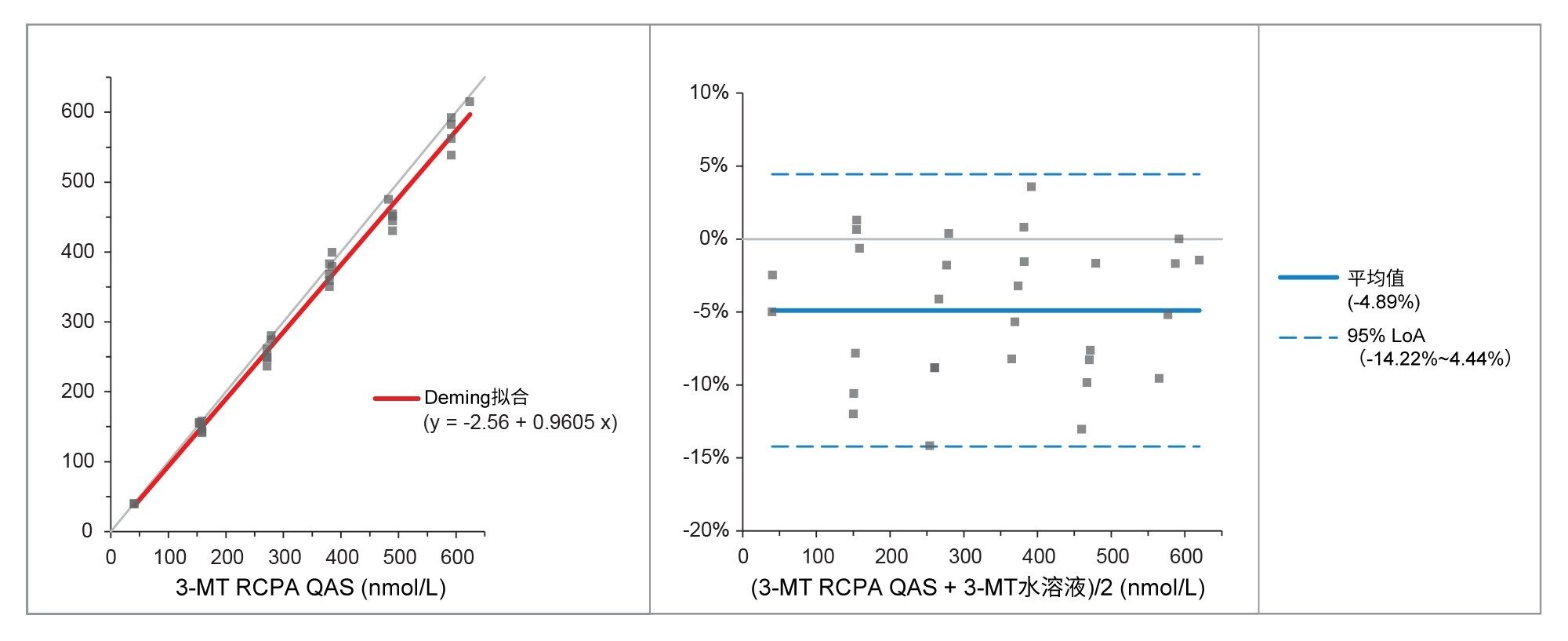 图5.比较实测值与RCPA QAS值得出的3-甲氧酪胺Deming拟合图和Bland Altman图
图5.比较实测值与RCPA QAS值得出的3-甲氧酪胺Deming拟合图和Bland Altman图
结论
本研究开发出一种用于分析人尿液中变肾上腺素、去甲变肾上腺素和3-甲氧酪胺的临床研究方法,该方法具有以下性能特征:
- 在所有运行中,所有分析物的标准曲线相关系数(r2)均>0.99
- 批次内精密度和批次间精密度结果为CV≤10.7%。
- 在进样高浓度样品之后立即进样空白样品时,未观察到明显的残留
- 将采集自6个供体的样品的实测浓度计算值与对照样品相比较,几乎没有观察到离子抑制效应
- 该临床研究方法与RCPA质量保证计划相比具有良好的一致性
720008227ZH,2024年2月