Medicinal herbs, including those used in Traditional Herbal Medicine (THM) or Traditional Chinese Medicine (TCM), have been used for many thousands of years. Recently their popularity in Western countries has burgeoned and there has been a shift in the way they are perceived; from alternative therapies to complementary medicines. Hence there is an increasing demand for THM products to meet and maintain stringent international quality standards of botanical, chemical, and clinical aspects.
Pharmaceutical companies are keen to identify single bioactive compounds that are extracted from THMs. However, the multi-component and synergistic nature of THMs means that it is beneficial to analyze complex extracts. In addition, it is desirable to generate a fingerprint, or a profile, of a given herb/THM to enable differentiation from similar plants, identification of impurities, or a comparison to determine differences occurring due to harvesting times, etc.
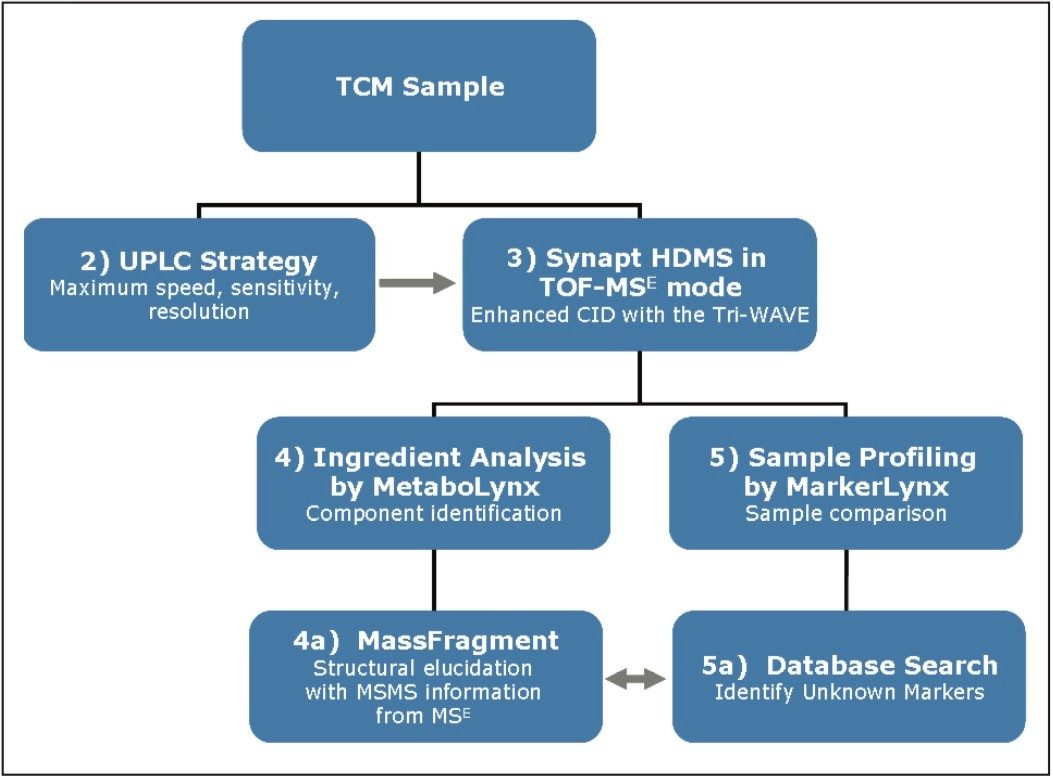
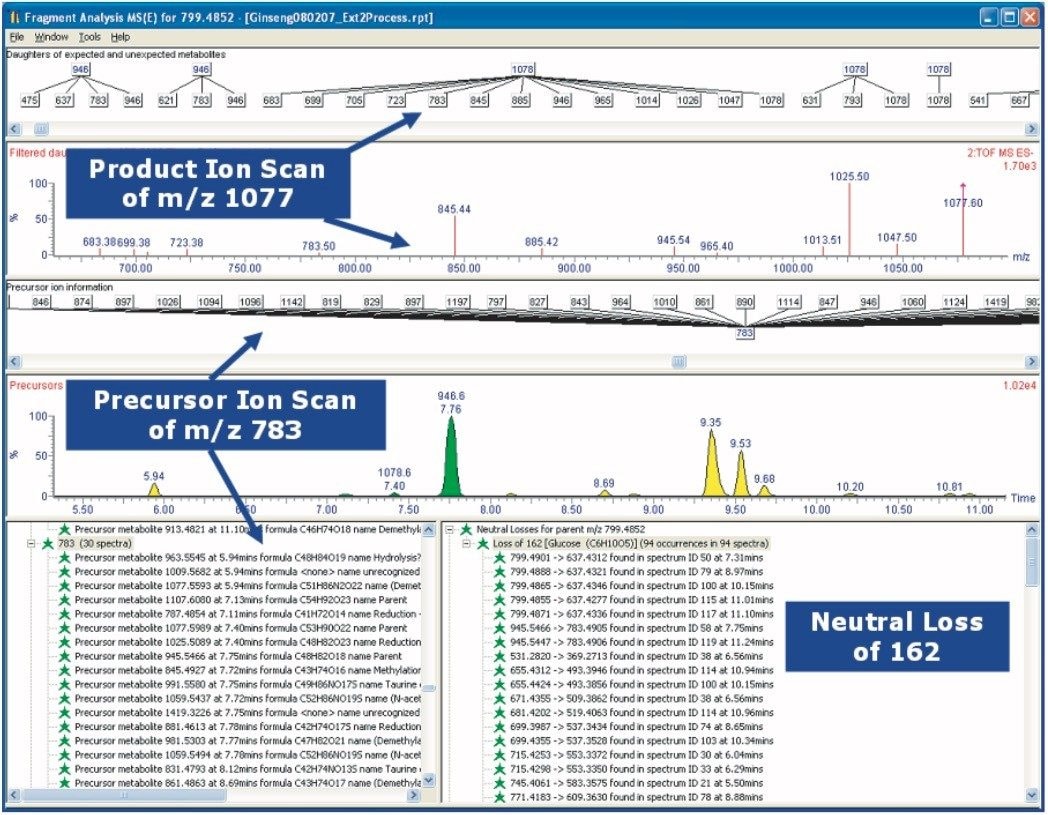
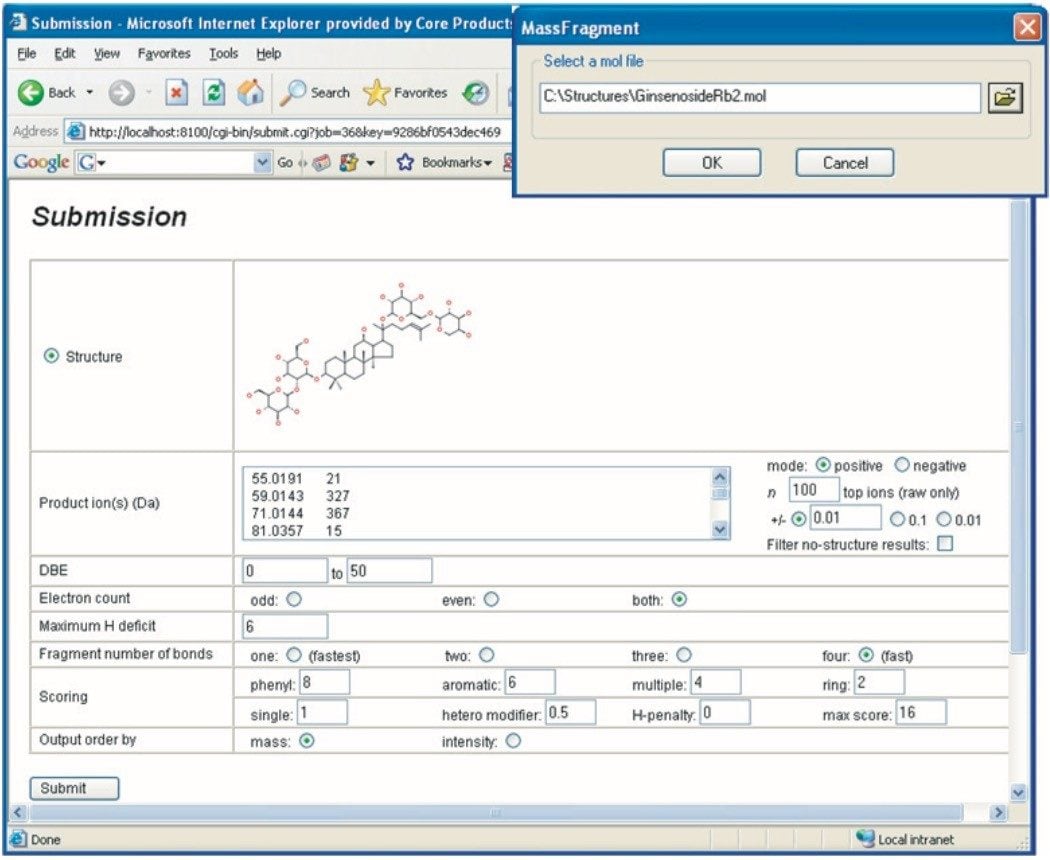
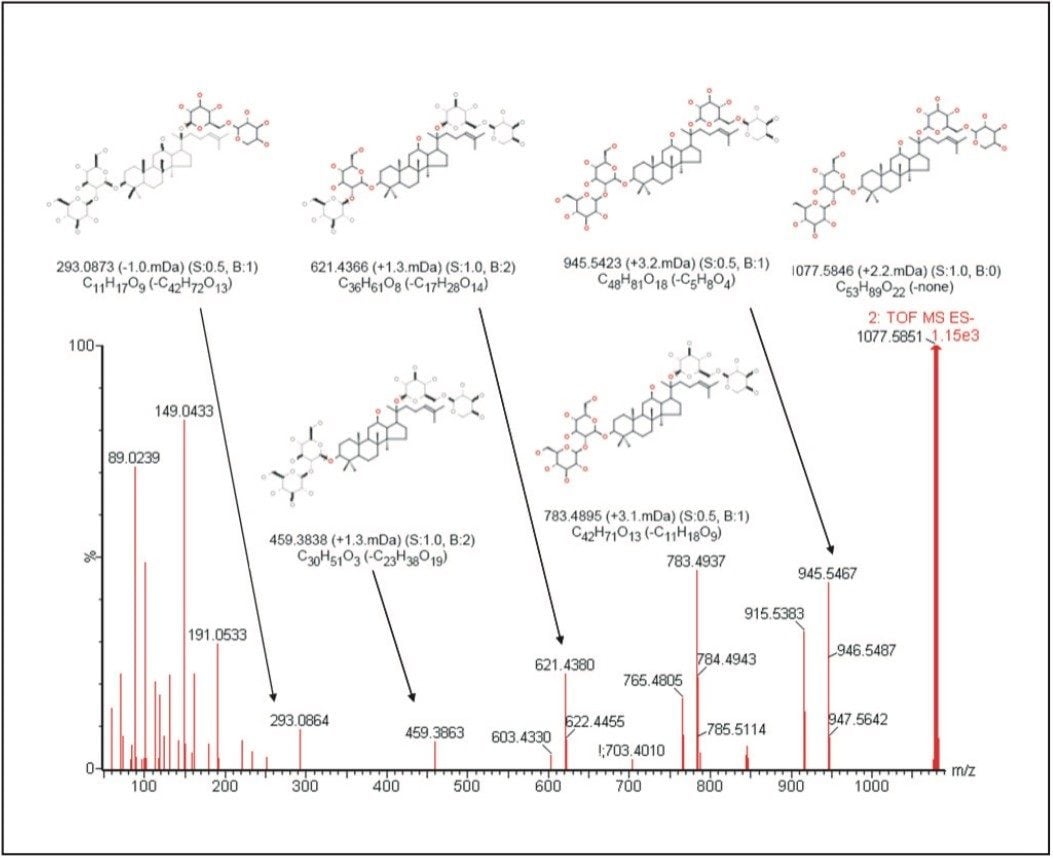
THM samples frequently contain hundreds or even thousands of individual chemical entities present in a wide range of concentration levels - and so their analysis is a challenging task. Here we present a generic workflow (Figure 1) using the Waters ACQUITY UPLC and SYNAPT HDMS systems, with its mass spectrometer in time-of- flight (TOF) mode.
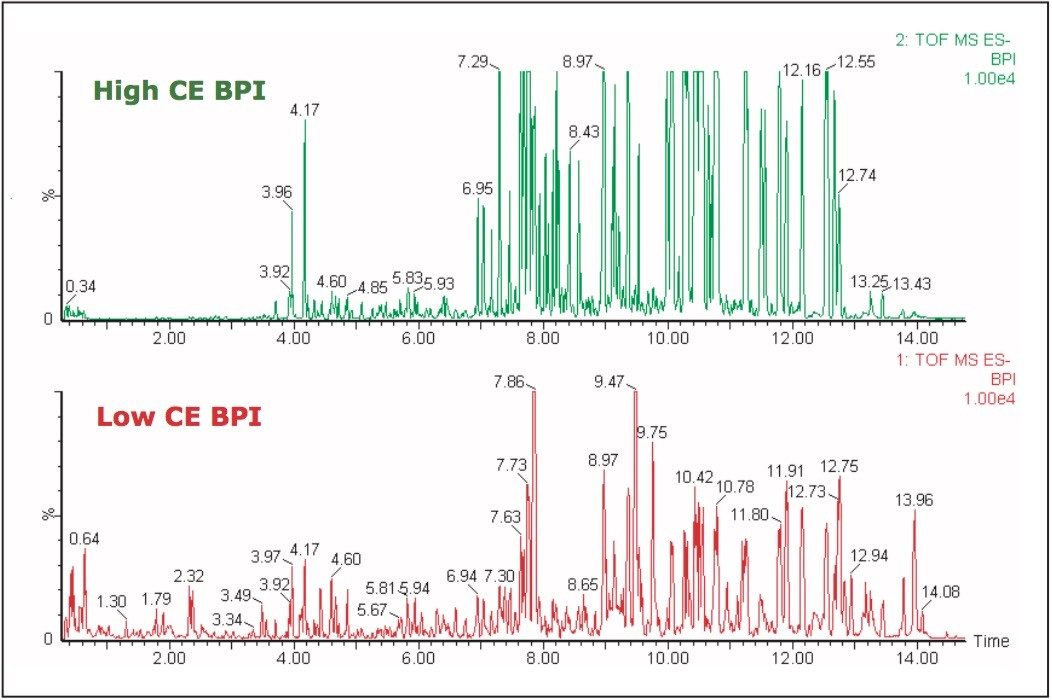
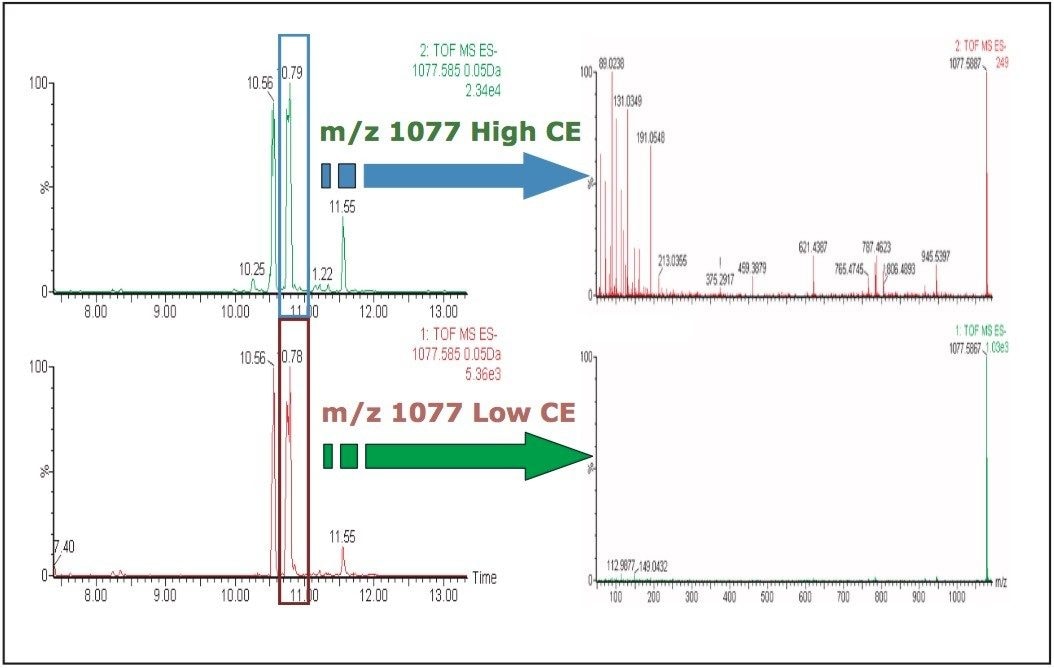
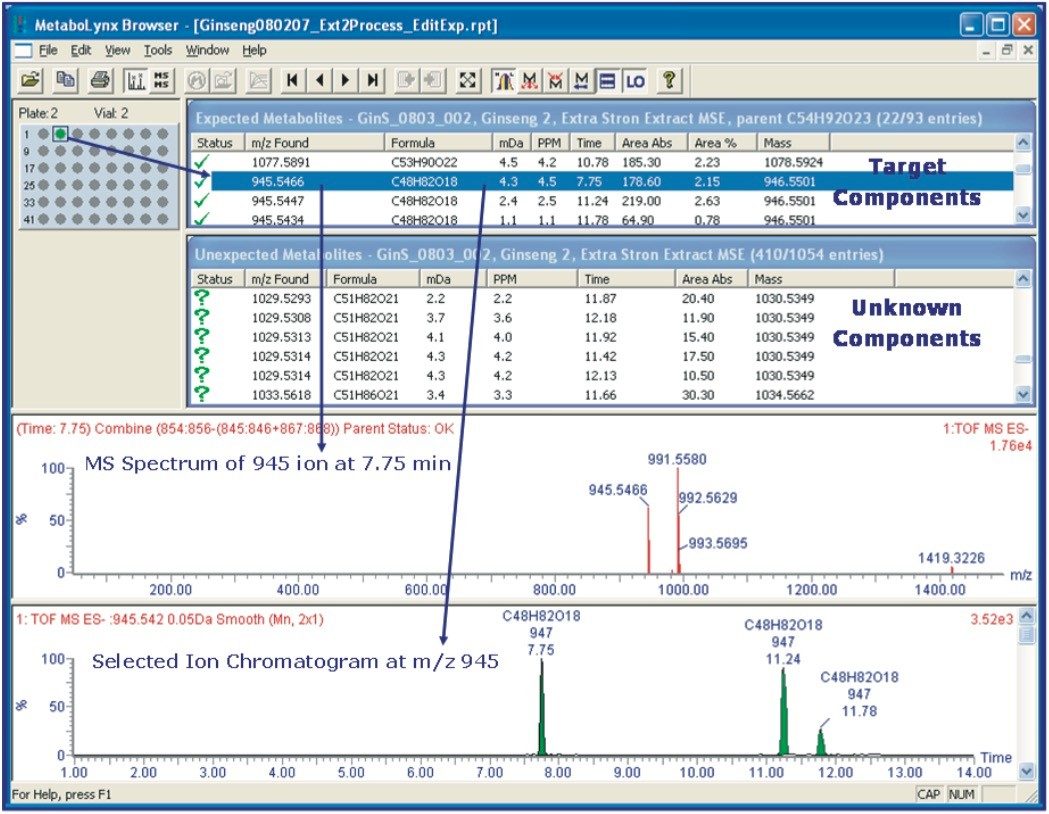
UPLC is a separation technique that offers high resolution, excellent sensitivity, and enables faster analyses. Data generated from MSE experiments performed on the SYNAPT HDMS System provides accurate mass information used for predicting the elemental composition of the chemical entities, and fragment ion data for structure confirmation in the absence of pure standards. This UPLC-HDMS system configuration makes analysis of these types of complex samples much easier.