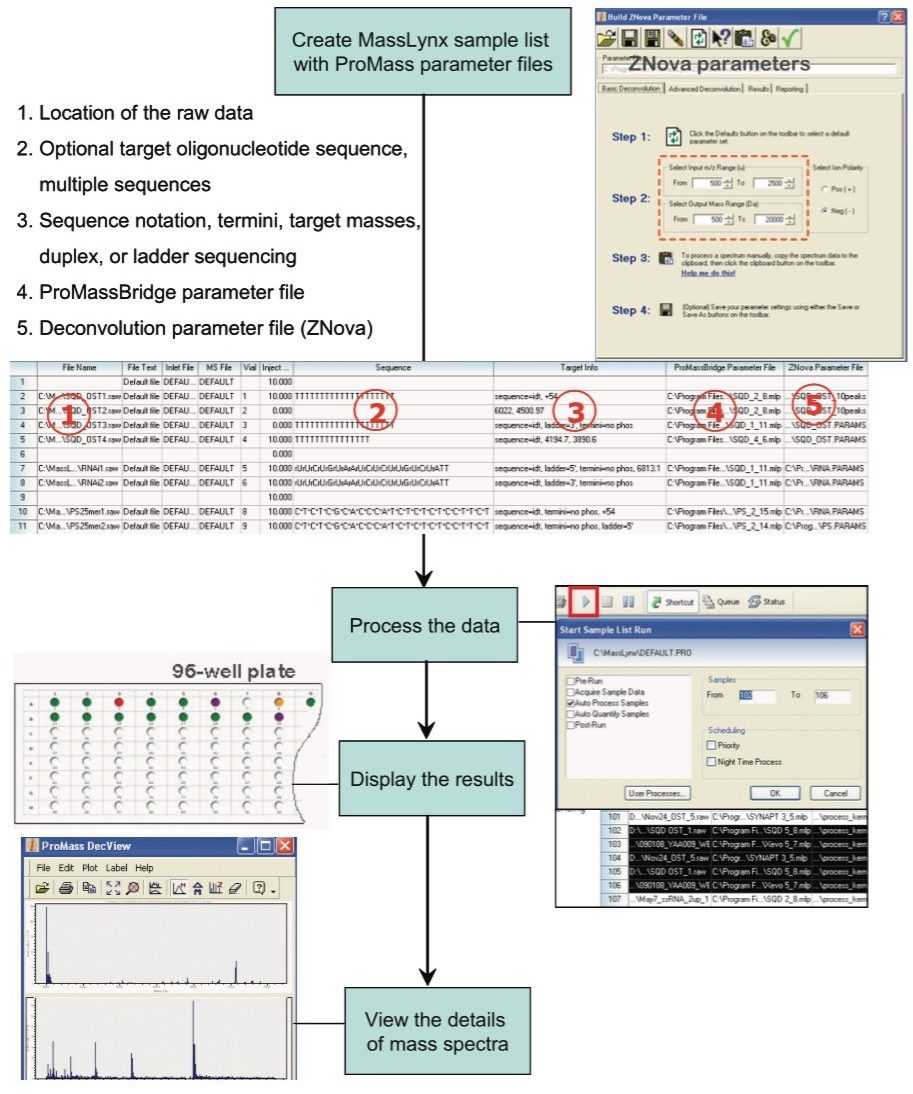
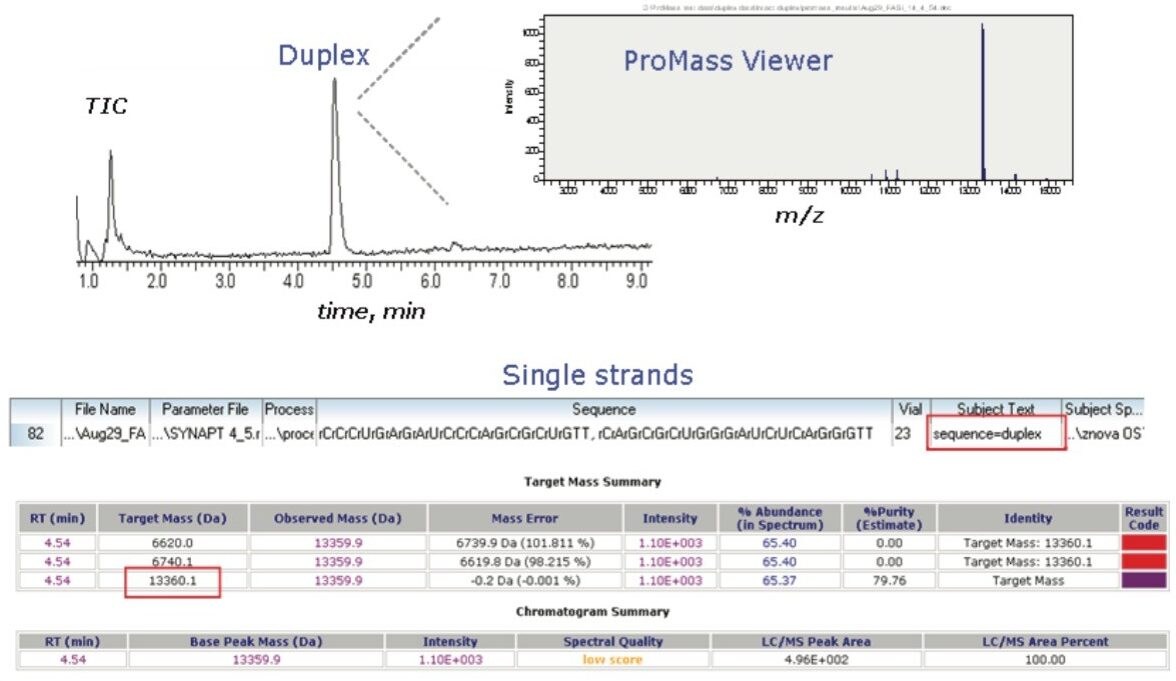
In the sample set, the user has the option to define the analyte(s) of interest either as target mass, sequence, or combination of both. A variety of nucleotides modifications is supported, such as DNA, RNA, LNA, 2'-O-methylation, 2'-fluorination, or phosphorothioate modificaton. The user can specify any modification according to the Integrated DNA Technology (IDT) notations, or define custom modifications. The target information may include the mass of the targeted product, the adducts, termini modifications, and a request for ladder sequencing or processing of a duplex. In high-throughput analysis mode, the user can display data summary as a 96-well plate with color-coded entries
(Figure 3). The colors represent the result of the targeted mass(s) search. It is offered in interactive format to allow navigation through the chromatograms and the details of mass spectra. There is a general chromatographic summary listing the peaks assigned within the retention time range defined in ProMassBridge parameter file. The mass spectra corresponding to each chromatographic peak can be viewed in detail in the ProMass Viewer window.
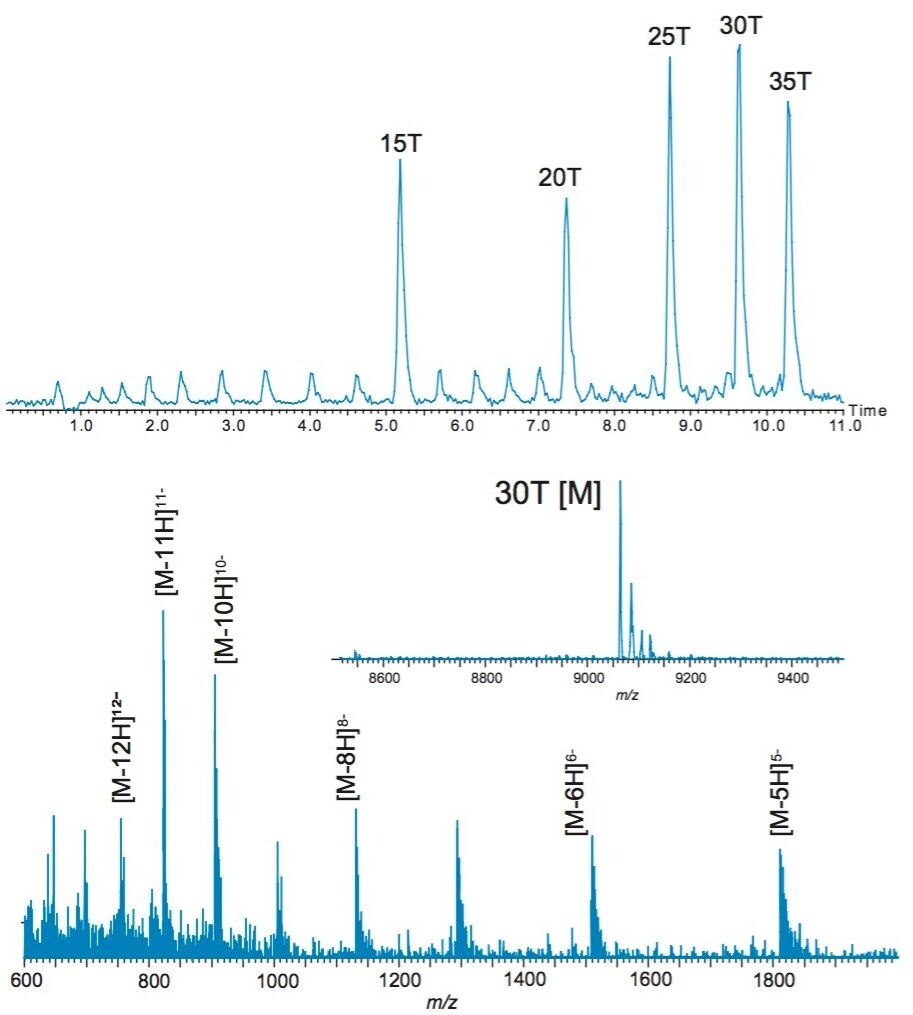
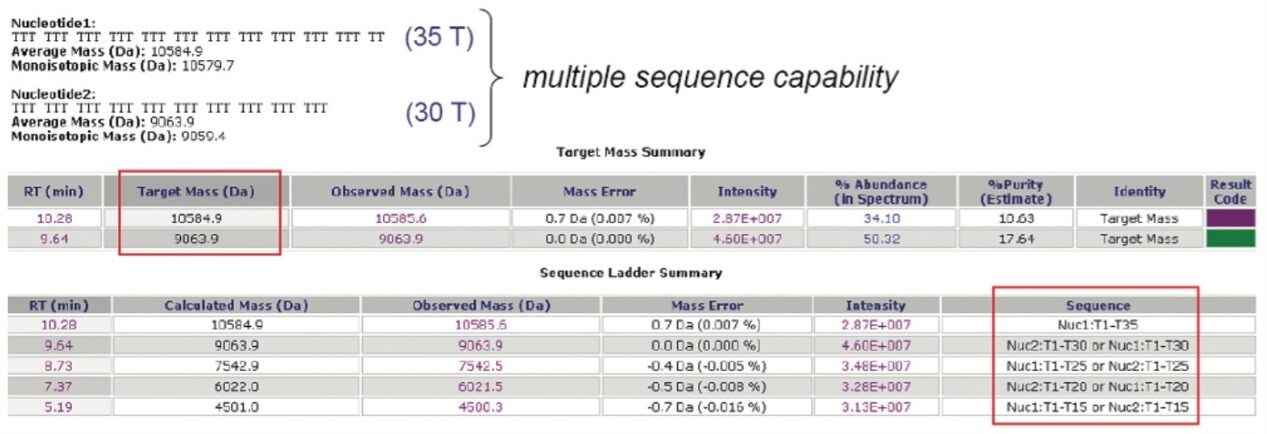
In order to test the correctness of ProMass for MassLynx, the data of OST standards acquired on an ACQUITY SQD single quadrupole mass spectrometer were processed (Figure 2). Multisequence capability was investigated using 30 T and 35 T OST homologs as two targeted mass searches. Generally, more than two sequences can be entered. The sequences of both homologs were supplied, and the ProMass for MassLynx calculated the theoretical masses and searched for the corresponding values within the specified time range.
The deconvoluted mass of 30 T species resulted in 0.0 Da mass error, and was marked in green, meaning that the target mass found in chromatogram is the most abundant component within the suggested 0.02% masserror tolerance. The 35 T homolog was marked in purple, which happens when other components present in the spectrum have greater than 30% abundance. The deconvoluted masses of the failed sequences of the targeted 35 T oligonucleotide (30 T, 25 T, 20 T, 15 T) were reported in the sequence ladder summary. ProMass for MassLynx successfully identified the targeted masses and picked the correct picks as truncated sequences. The results correlated with manually deconvoluted masses of OST homologs (Figure 1).
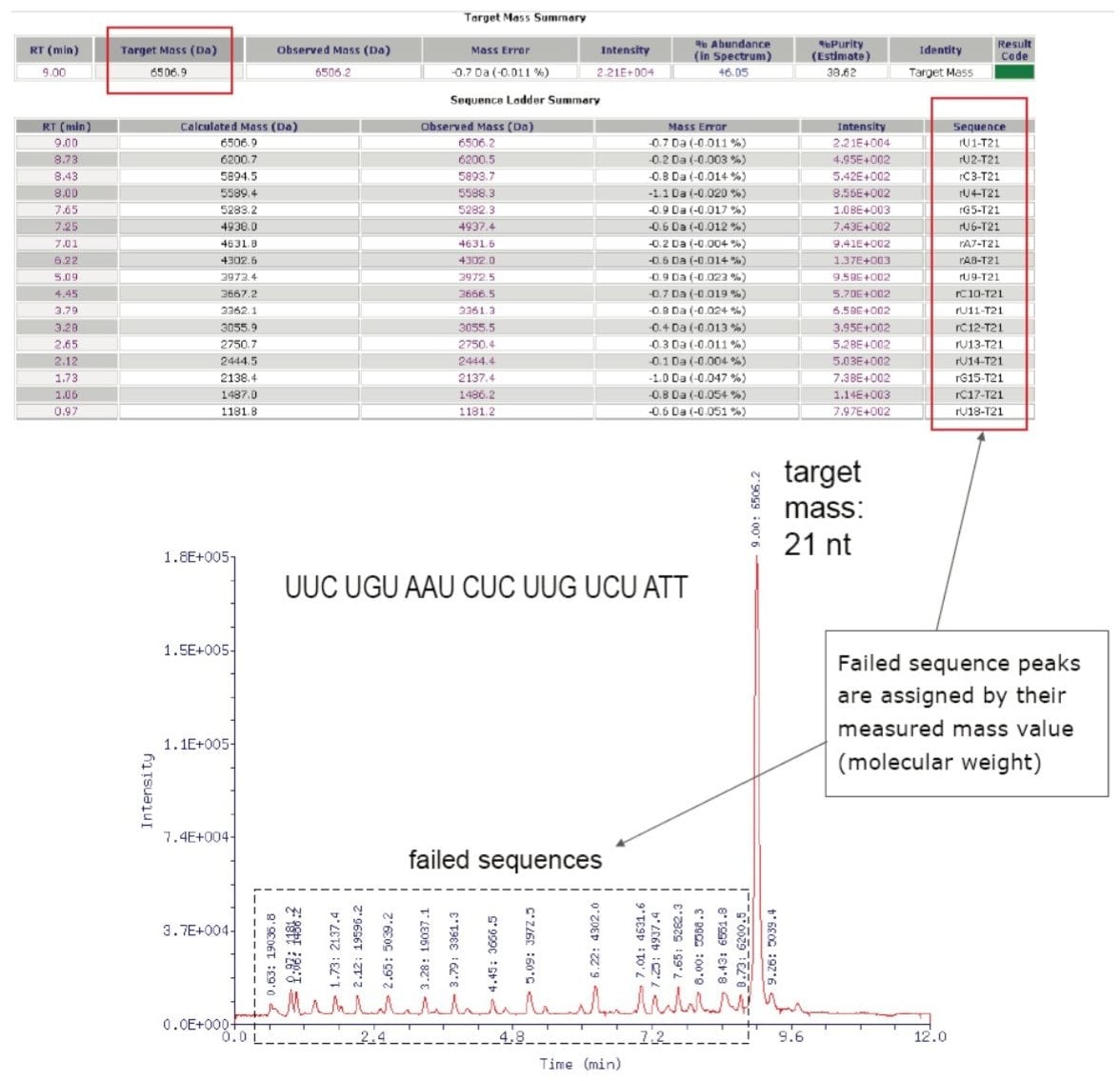
To process the OST chromatogram, the reporting option in the ZNova parameter file was chosen to define the threshold of the chromatographic peaks selected for deconvolution so only the major homologs of the truncated sequences were taken into consideration (Figure 4). Assignment of the entire chromatogram is possible, as was demonstrated in Figure 4 in an example of 21-mer RNAi data obtained on an SYNAPT HDMS instrument. The molecular weight of the intact molecule was calculated based on the provided sequence and deconvoluted by ProMass for MassLynx with 0.011% mass error. In the Target Info column the request for the ladder sequencing was denoted as “ladder=5”.
Generally, the search for the truncated sequences from either 5' or 3' ends is available as are internal cleavages. As a result, 17 truncated sequences were reported in the sequence ladder summary. The structures of these sequences are reported in the summary table rather than assigned directly on the chromatogram.
Using the automatic peak deconvolution, most of the 21-mer sequence of RNAi was confirmed in a matter of a few minutes. The speed of UPLC separation coupled with automated ProMass for MassLynx processing makes this method fast and efficient. The interactive interface covers the vast range of the user's needs: from a quick summary report to the details of the mass spectra.