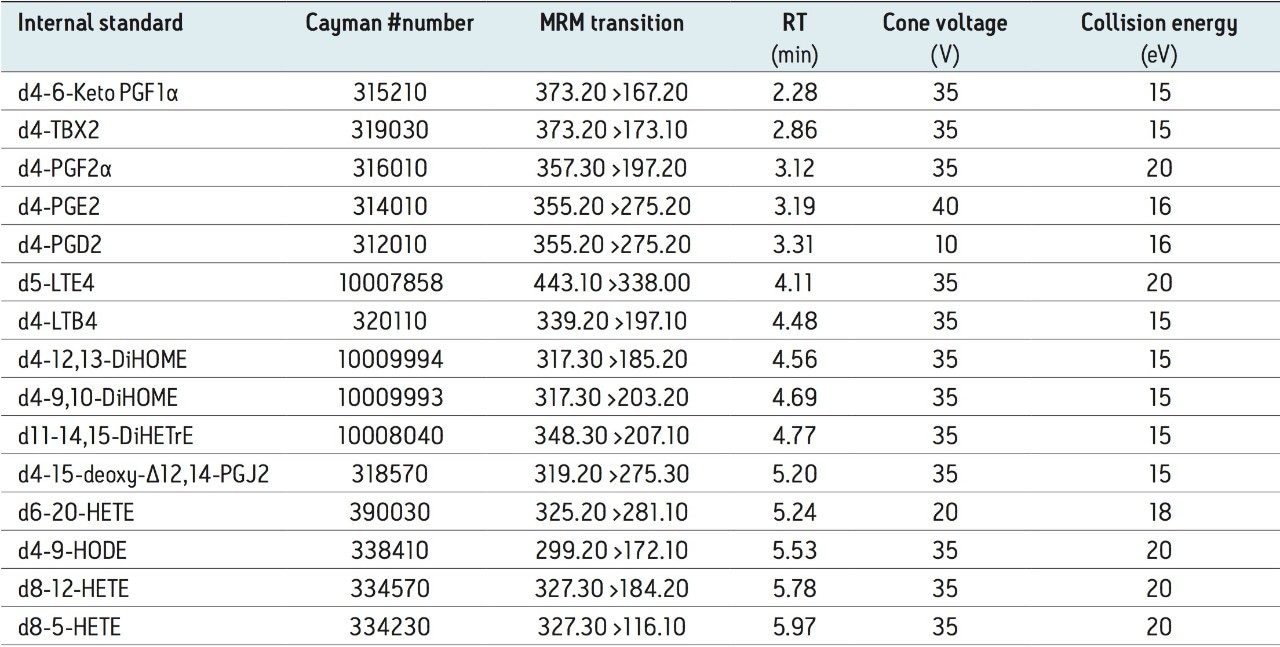
Oxylipins are signaling lipids that play prominent roles in the physiological regulation of many key biological processes, such as the relaxation and contraction of smooth muscle tissue, blood coagulation, and most notably inflammation. Alterations in oxylipin pathways have been associated with response to cardiovascular diseases, host defense, tissue injury and surgical intervention. The ability to semi-quantitatively profile a wide range of oxylipin in plasma samples could help our understanding of their roles in health and disease, as well as serve as biomarkers for disease diagnosis or prognosis.
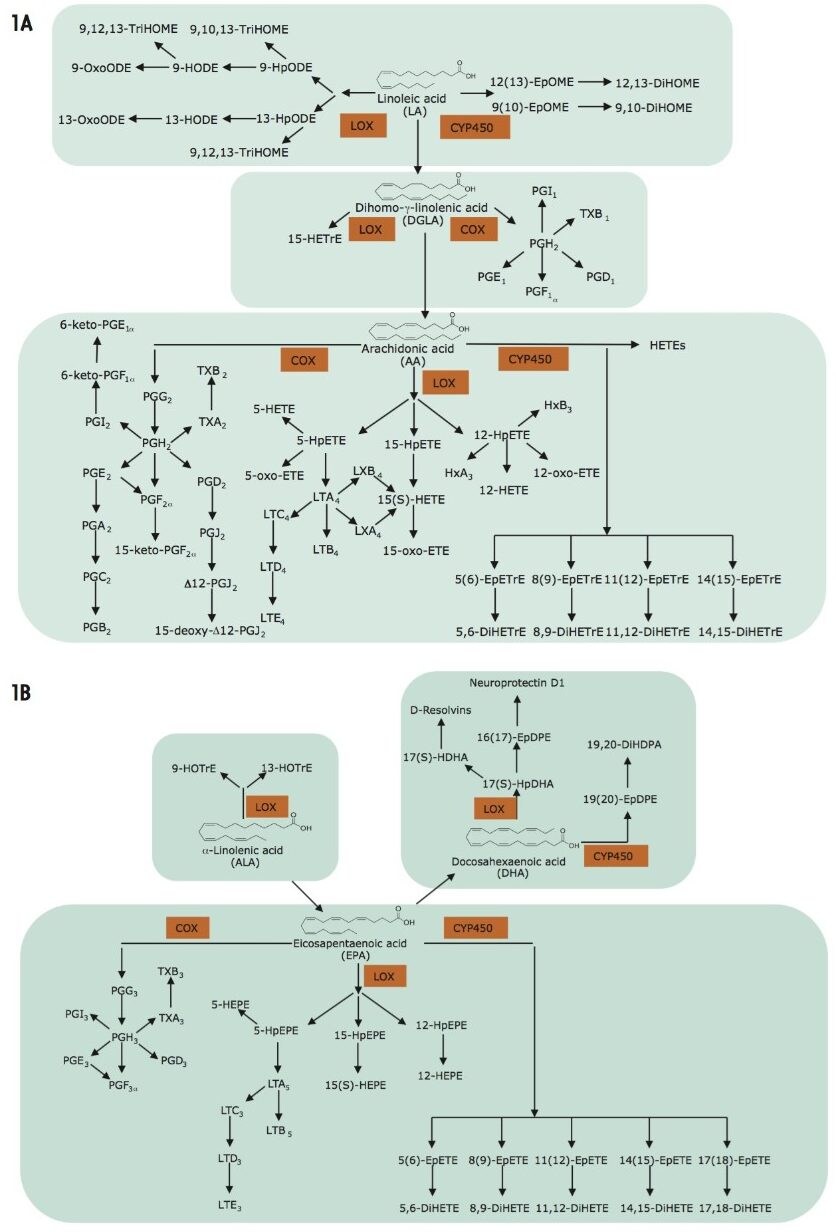
Oxylipins are produced via enzymatic (e.g., mono- or dioxygenase-catalyzed) or non enzymatic oxygenation of an array of both omega-6 polyunsaturated fatty acid substrates (e.g., linoleic acid, dihomo-γ-linolenic acid, adrenic acid and arachidonic acid) and omega-3 polyunsaturated fatty acid substrates (α-linolenic acid, acid, eicosapentaenoic acid, and docosahexaenoic acid) (Figure 1A and 1B). Three major enzymatic pathways are involved in their generation: cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP). These pathways are important drug targets for multiple diseases (Figure 1A and 1B).
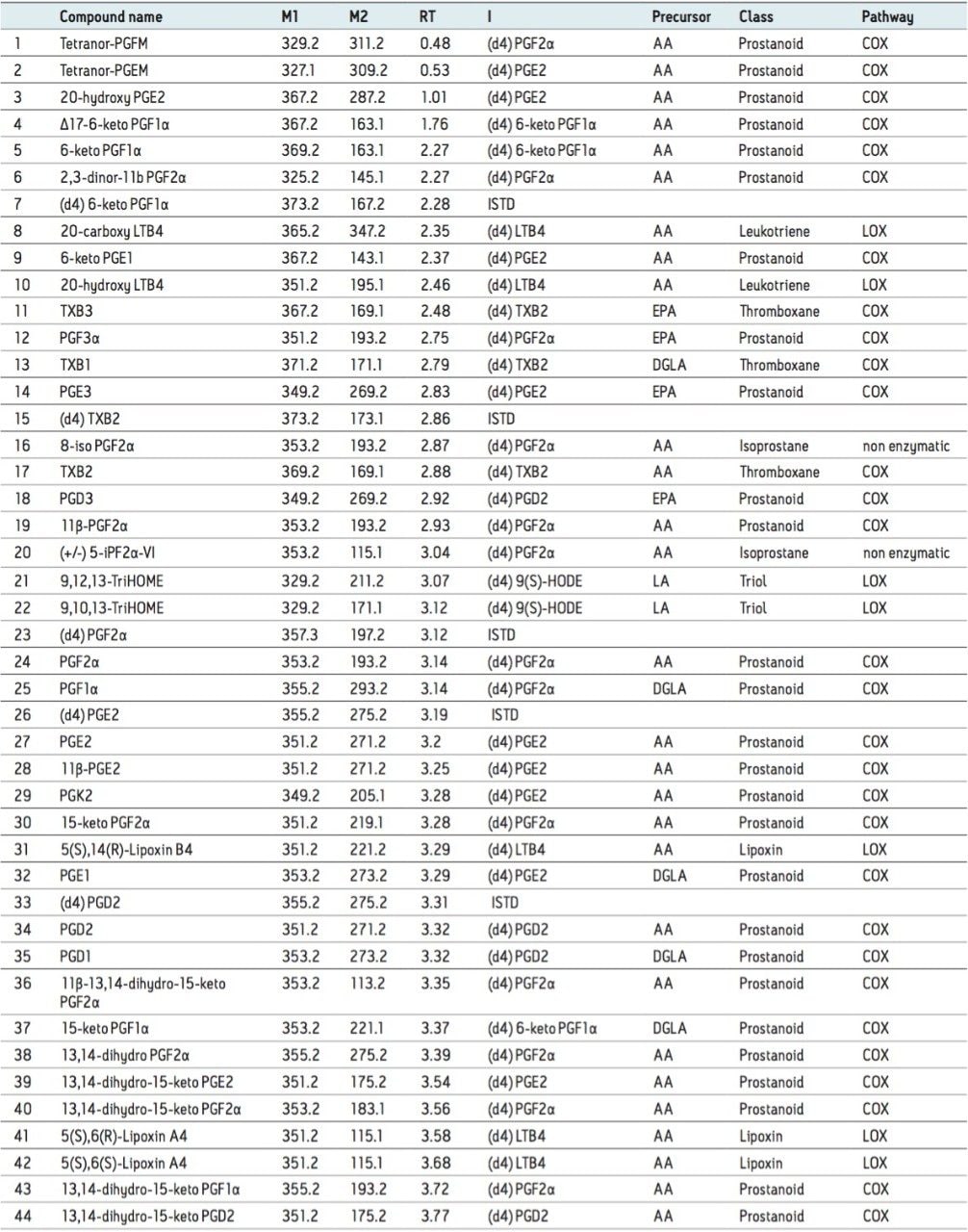
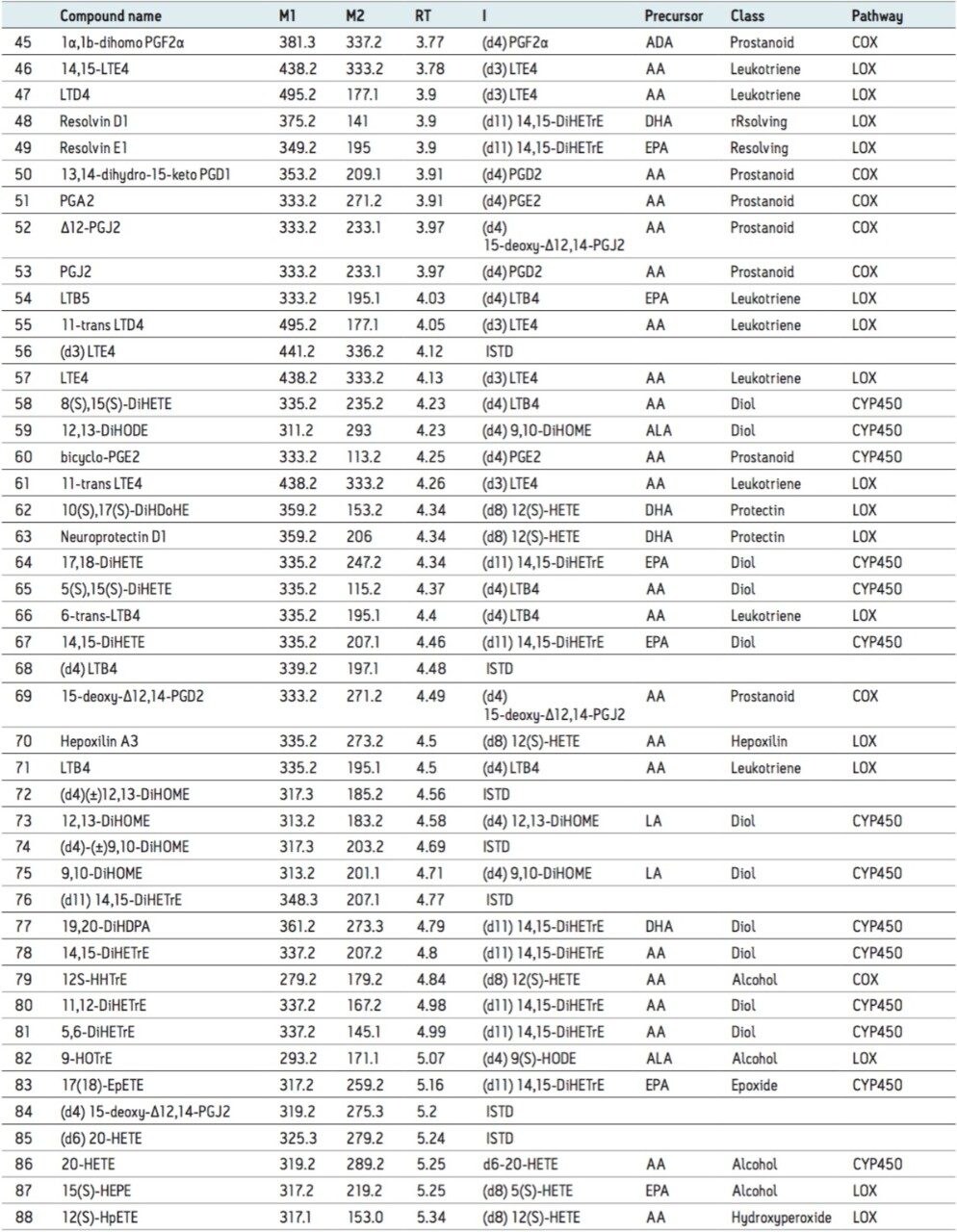
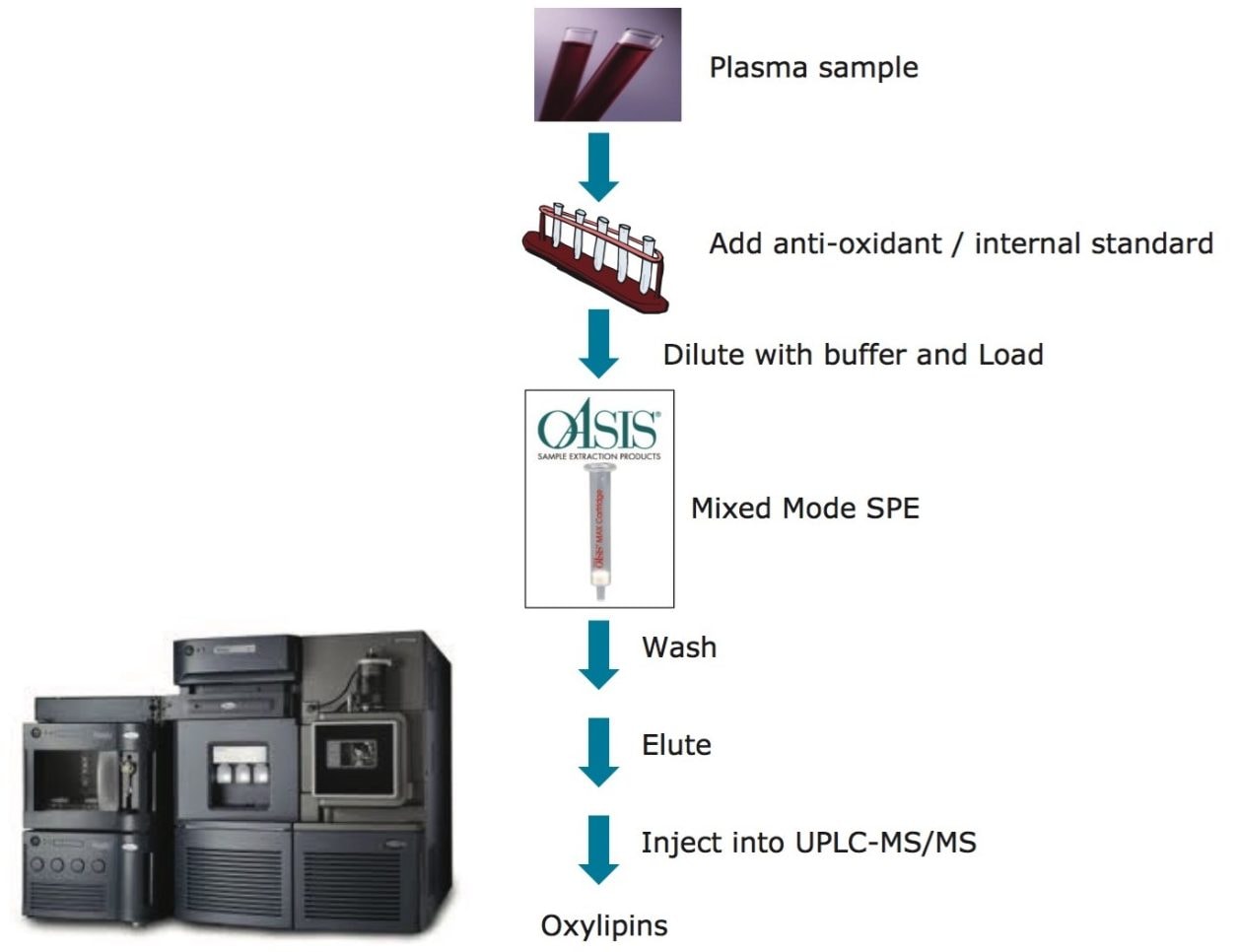
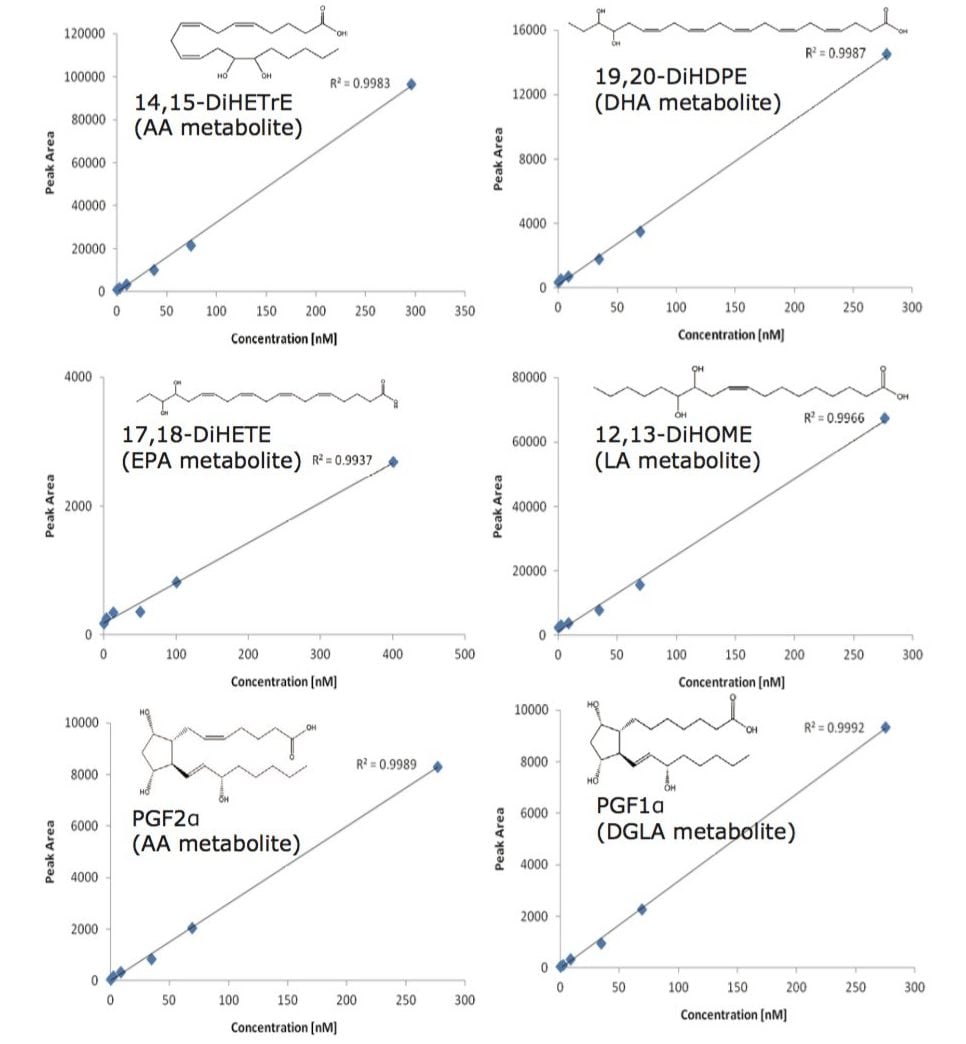
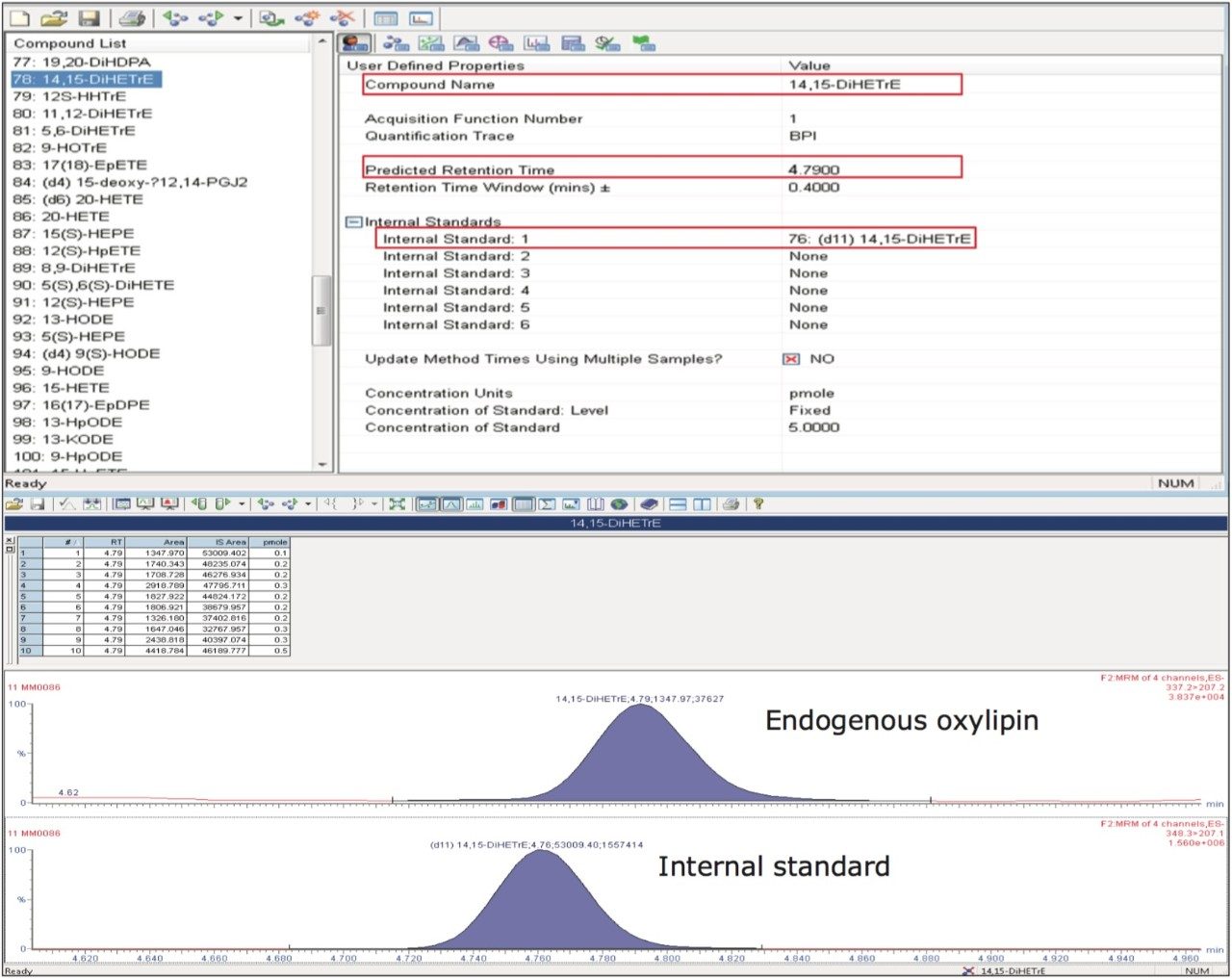
The main challenge for the measurement of oxylipins is the extremely low endogenous concentration of such lipid species and their limited stability. Furthermore, oxylipins are not stored in tissues but are formed on demand by liberation of precursor fatty acids from esterified forms. Lastly, the same fatty acid can be oxidized in different positions of its acyl chain leading to many isomeric species, each with specific metabolic actions. As a consequence, this requires a rapid, highly-sensitive, and specific analytical method.
Historically, measurements of oxylipins have been performed using radiometric and enzymatic immunoassays, which often lacked specificity and targeted only few compounds. GC-MS methodology has also been used, but this still requires multi-step procedures involving derivatization of the oxylipins to increase their volatility and stability.
Recently, various LC-MS methodologies have been described to monitor a broad range of low abundance oxylipins.1-5 In particular the method by Strassburg et al.2 reports on a wide range of oxylipins produced both enzymatically and non-enzymatically in human plasma. Although such methods are both sensitive and specific, there is an increasing demand for a comprehensive and high-throughput screening method to enable wide-ranging lipidomic studies.
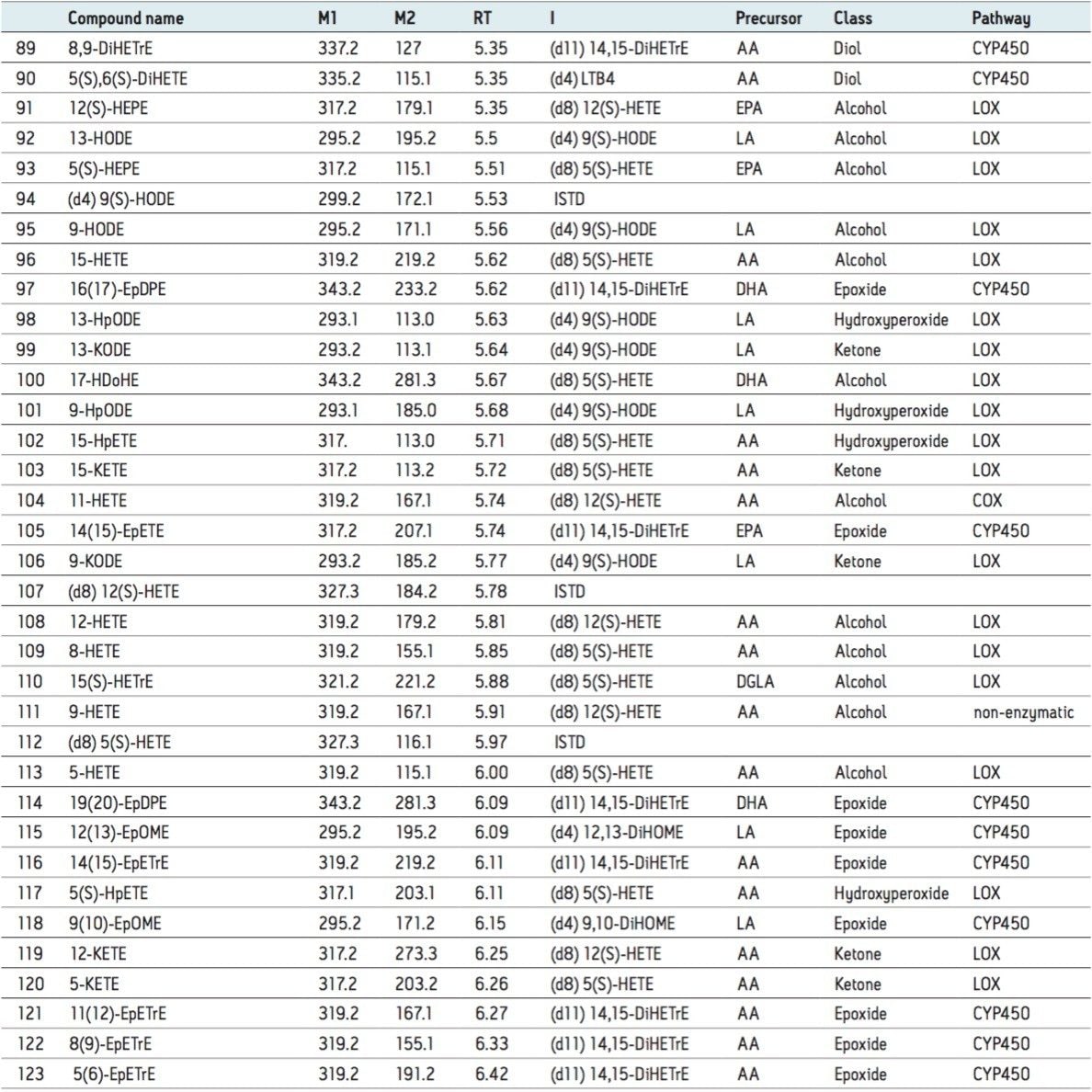
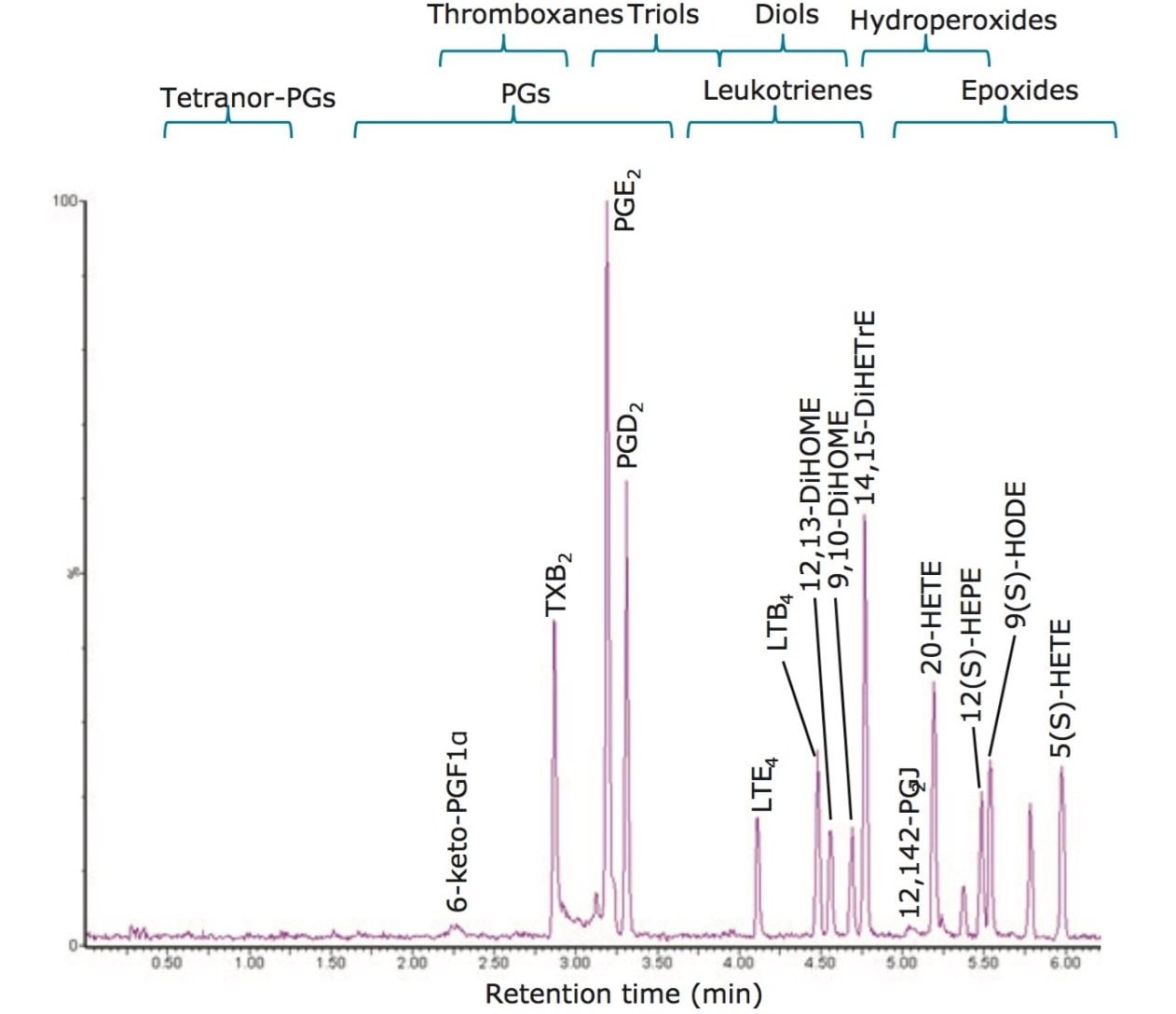
Here we report a high-throughput assay for the profiling of over 100 oxylipins, including prostaglandins, prostacyclines, thromboxanes, dihydroprostaglandins, and isoprostanes, in plasma samples.