Food colors are food additives that are used to make up for color losses following exposure to light, air, moisture, and variations in temperature, to enhance naturally occurring colors and to add color to foods that would otherwise be colorless or colored differently. Sudan dyes are not approved for use as colors in food in many countries including China, Australia, Canada, and Europe. For example, in the European Union, all additives, including colors, must be authorized and listed with conditions of use in the EU's ‘positive’ list contained in Regulation (EC) 1333/2008 and subsequent amendments.1 The presence of industrial dyes in food constitutes an adulteration of food products, since these substances are not authorized as food colors and their intake in food is a perceived risk to health. Azo dyes have been fraudulently and illegally used to enhance the visual surface color of spices and food.2 It has been estimated that the total cost of the recall of contaminated products in the UK in 2005, including loss of sales, destruction of goods, brand image damage, management time, and consultancy fees ran to over €200 million.
In the last decade, the number of reports of the presence of Sudan dyes has decreased substantially probably due to the increased monitoring of these compounds in spices. Nevertheless, such adulterations remain of concern to the consumer, food industry, and regulatory bodies in various parts of the world. European food manufacturers, distributors, and retailers have a duty to ensure that their products are compliant with EU regulations and demonstrate due diligence in their operations. Robust monitoring programs based on reliable detection methods are required to assure the food is free from harmful colors. Various analytical methods have been reported in the literature for the determination of dyes in food and beverage.3 Methods should cover those dyes frequently detected (e.g. RASFF) and included in any mandatory import testing controls, those dyes considered as a risk to health by EFSA’s Scientific Panel and those based upon industrial intelligence. All these dyes can be analyzed by LC-MS/MS, but not within 1 run, so the very polar dyes tend to be excluded. Dyes have been detected in a wide range of food ingredients and finished products, but typically spices (e.g. dried chili, chili products, and curry powder) and palm oil are the highest priority and were subject to increased level of official controls. As these compounds are banned, they have no maximum permitted limits. For practical purposes there tends to be a concentration below which no action is taken if adulterated is detected; e.g. from accidental contamination. The Standing Committee on the Food Chain and Animal Health, SCoFCAH, at a meeting held in Brussels on 23 June 2006, decided that in order to adopt a consistent approach to enforcement across Europe, applied an action limit of 0.5 mg/kg to illegal dyes in food ingredients such as spices and palm oil.4 Subsequently, this was implemented by competent authorities in the various Members States, including the FASFC (Federal Agency for the Safety of the Food Chain) in Belgium.
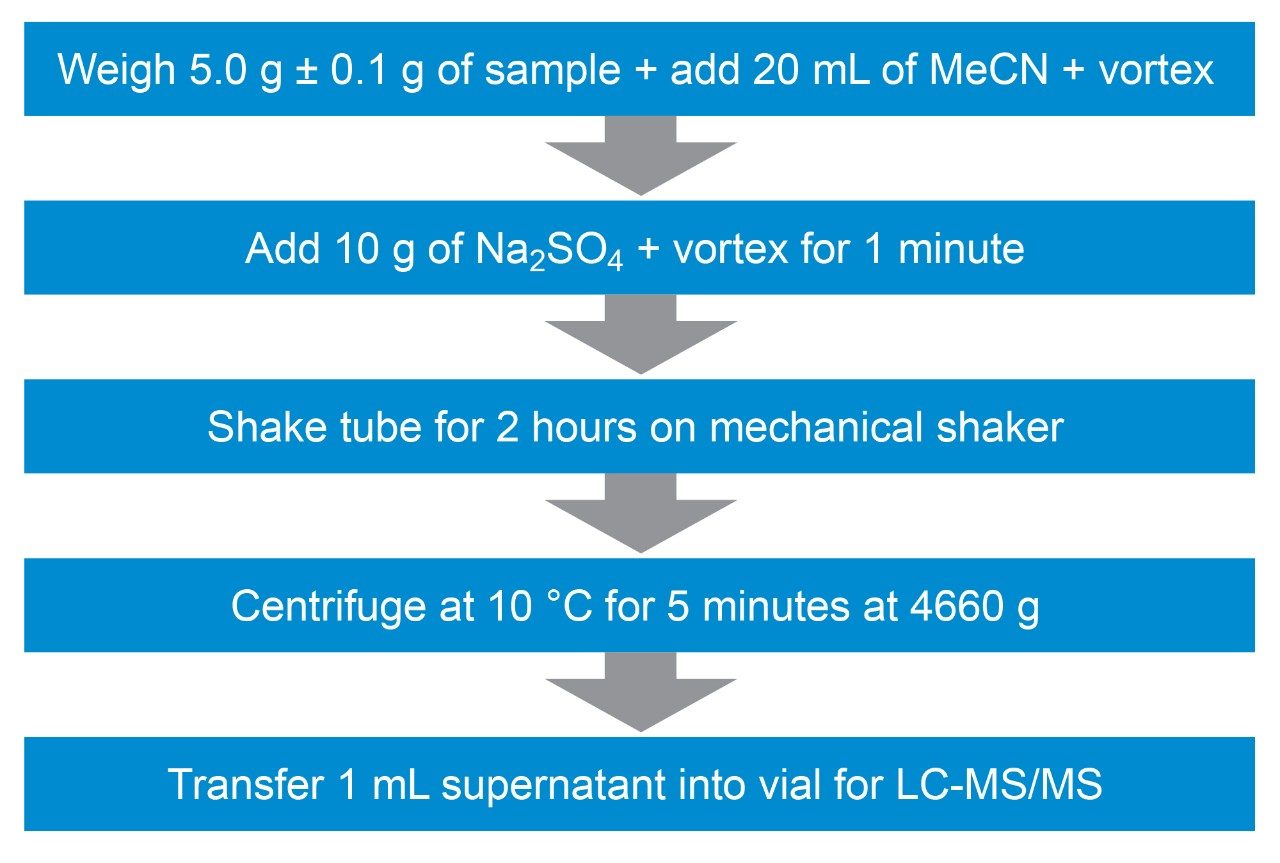
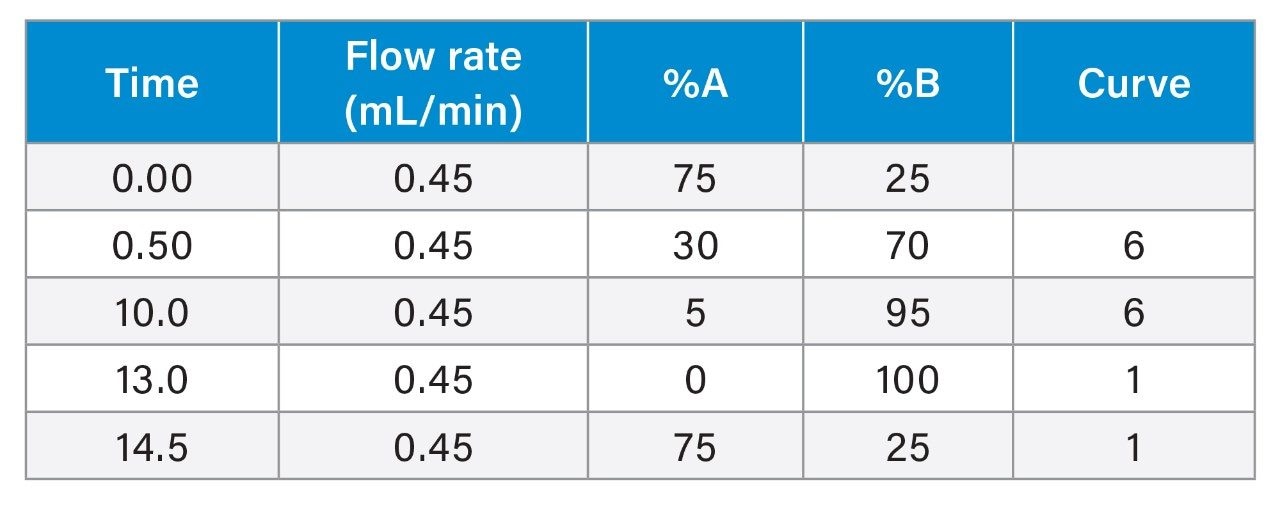
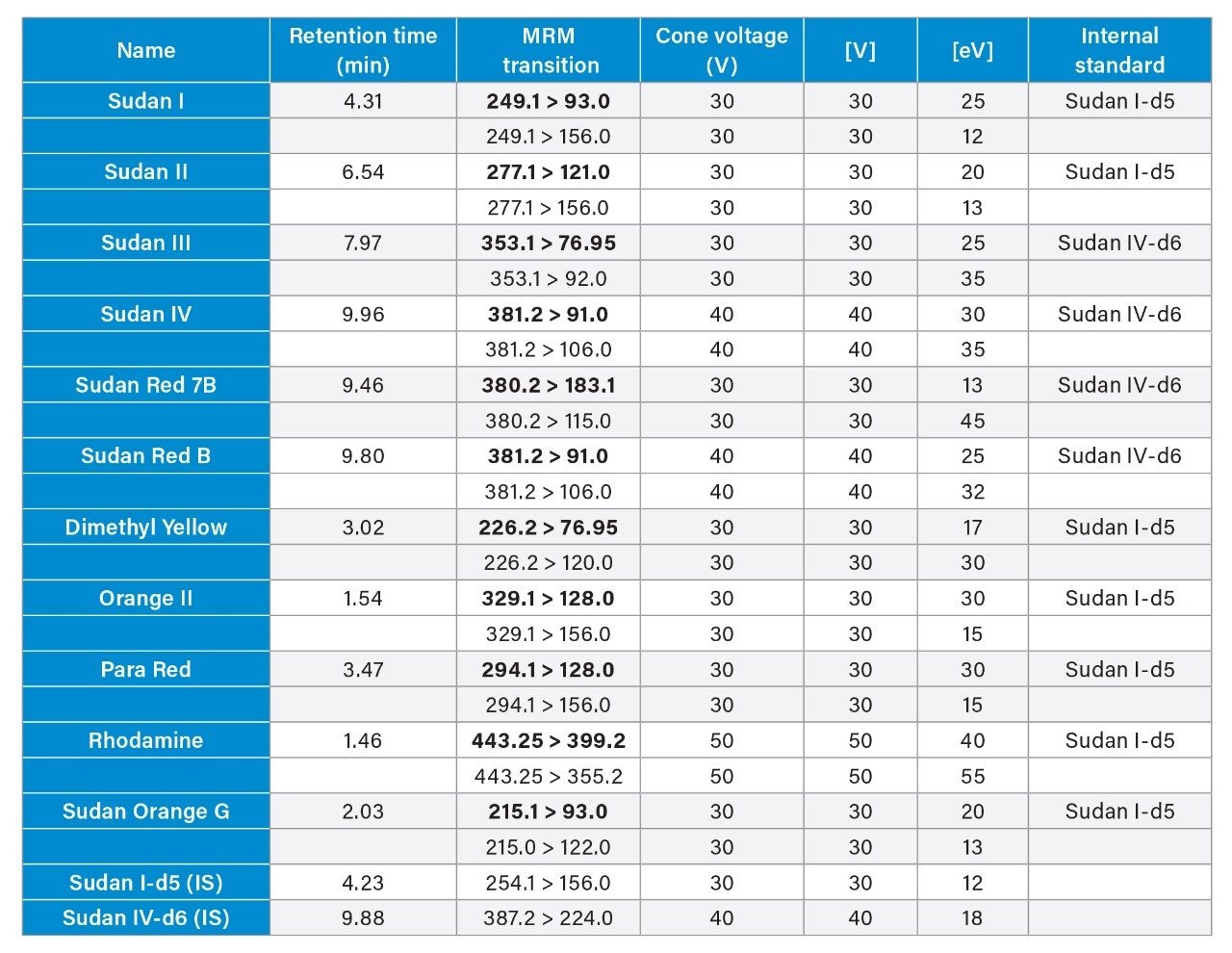
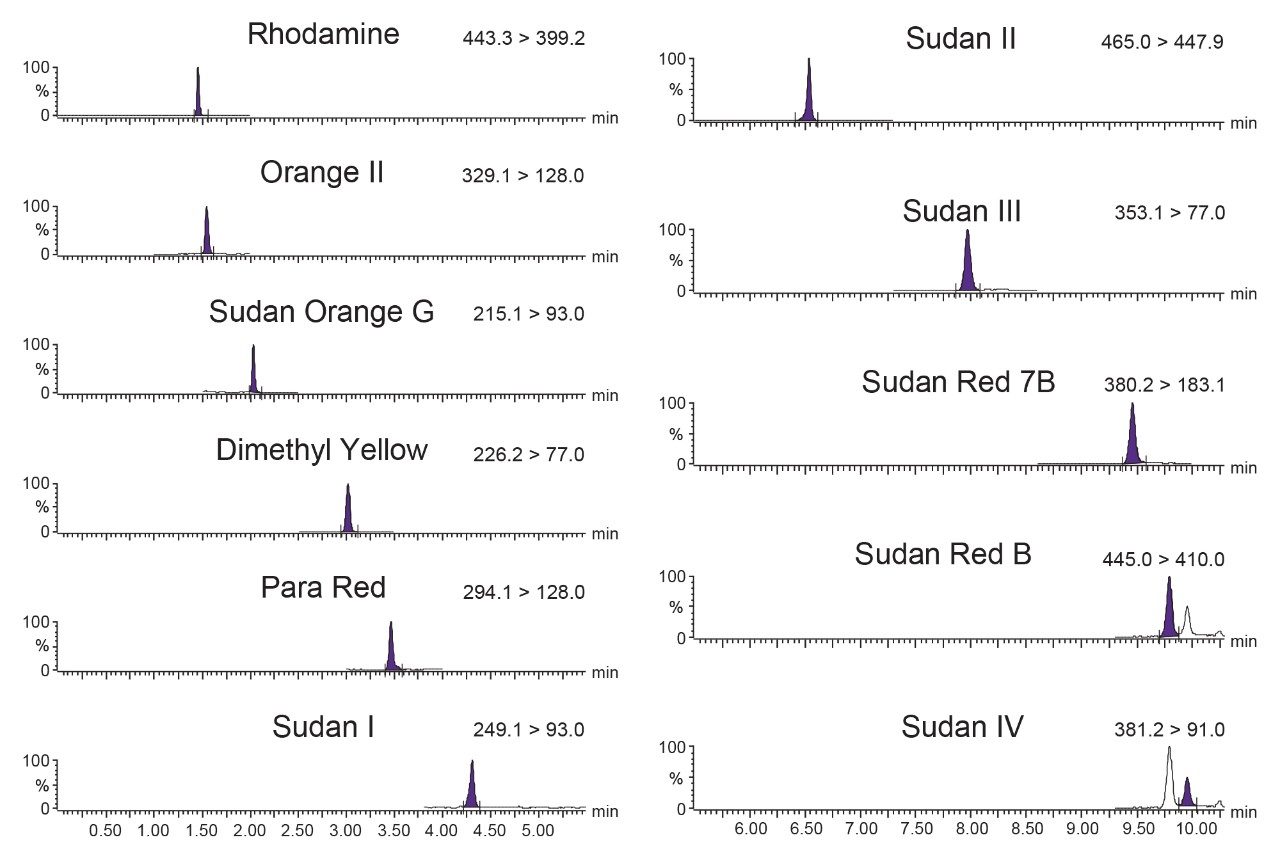
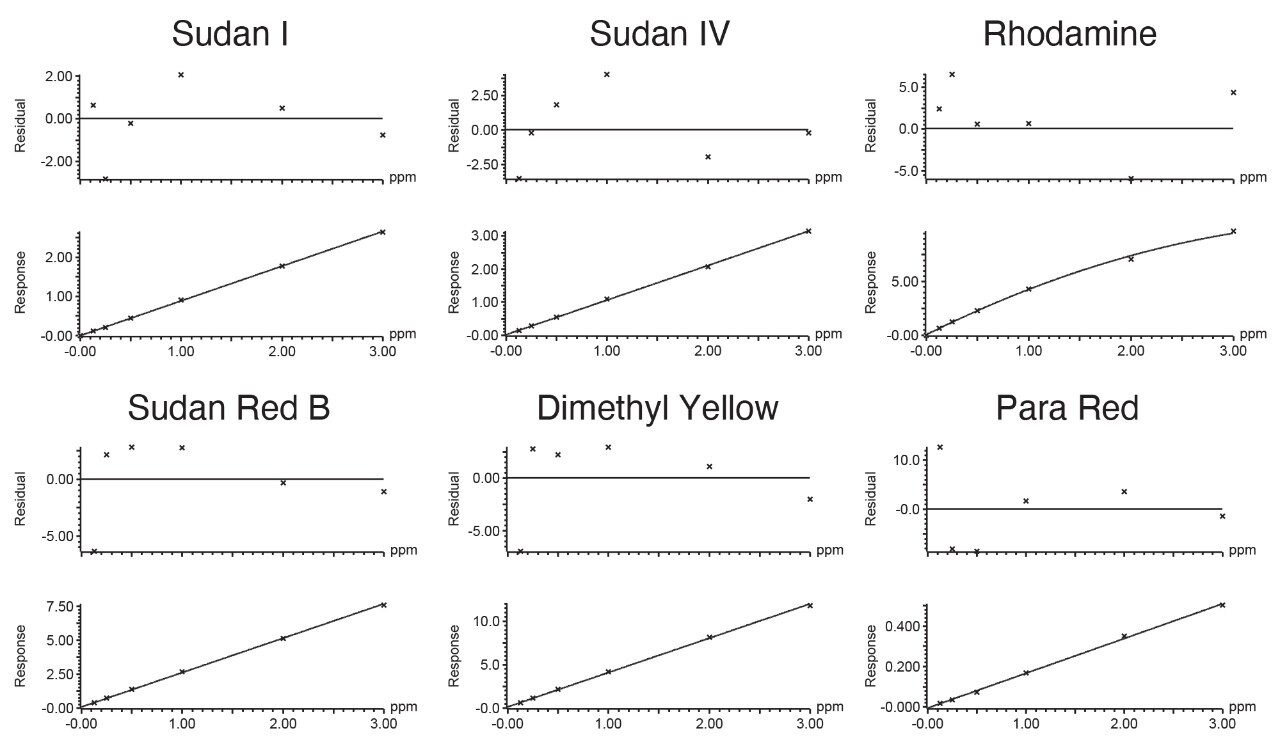
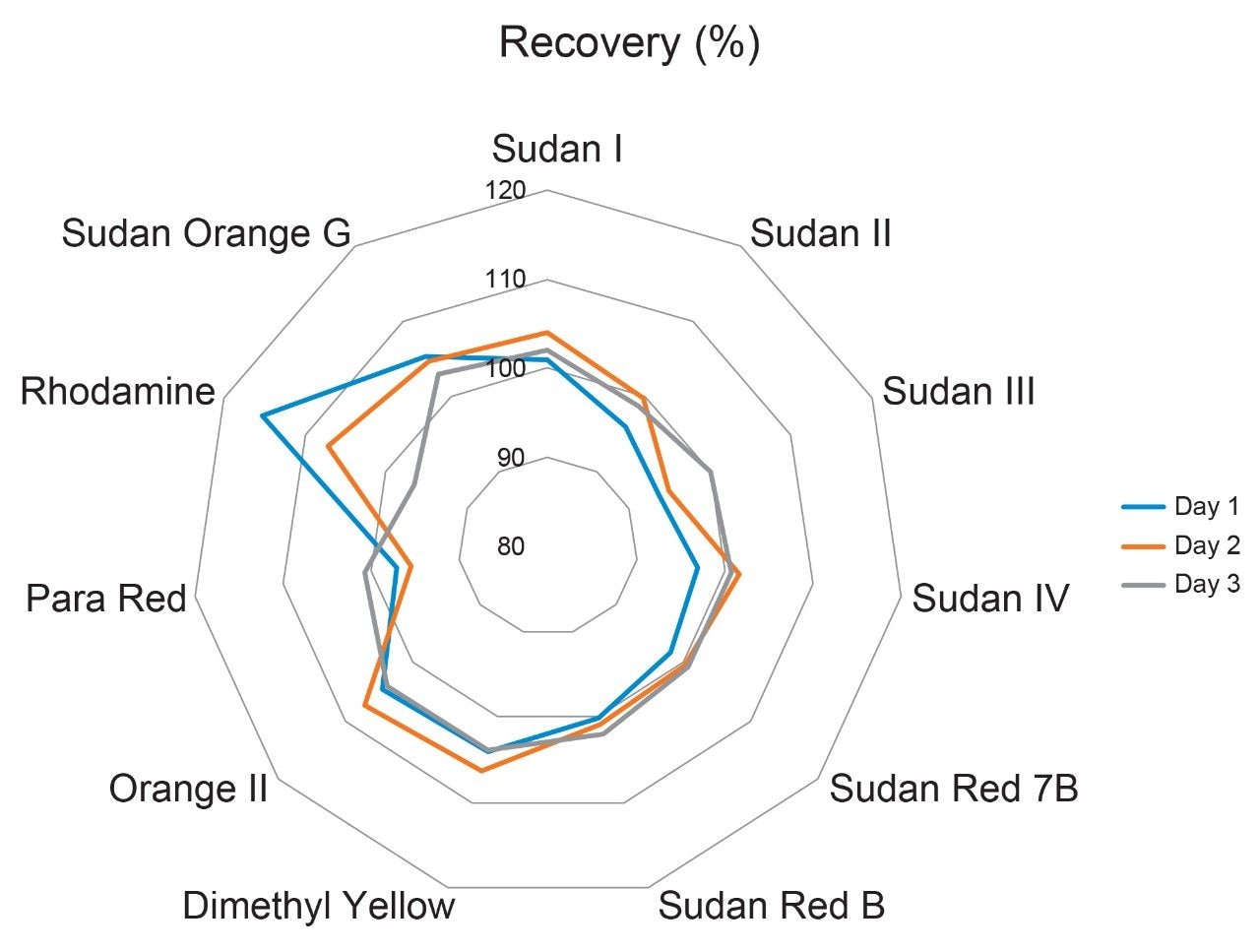
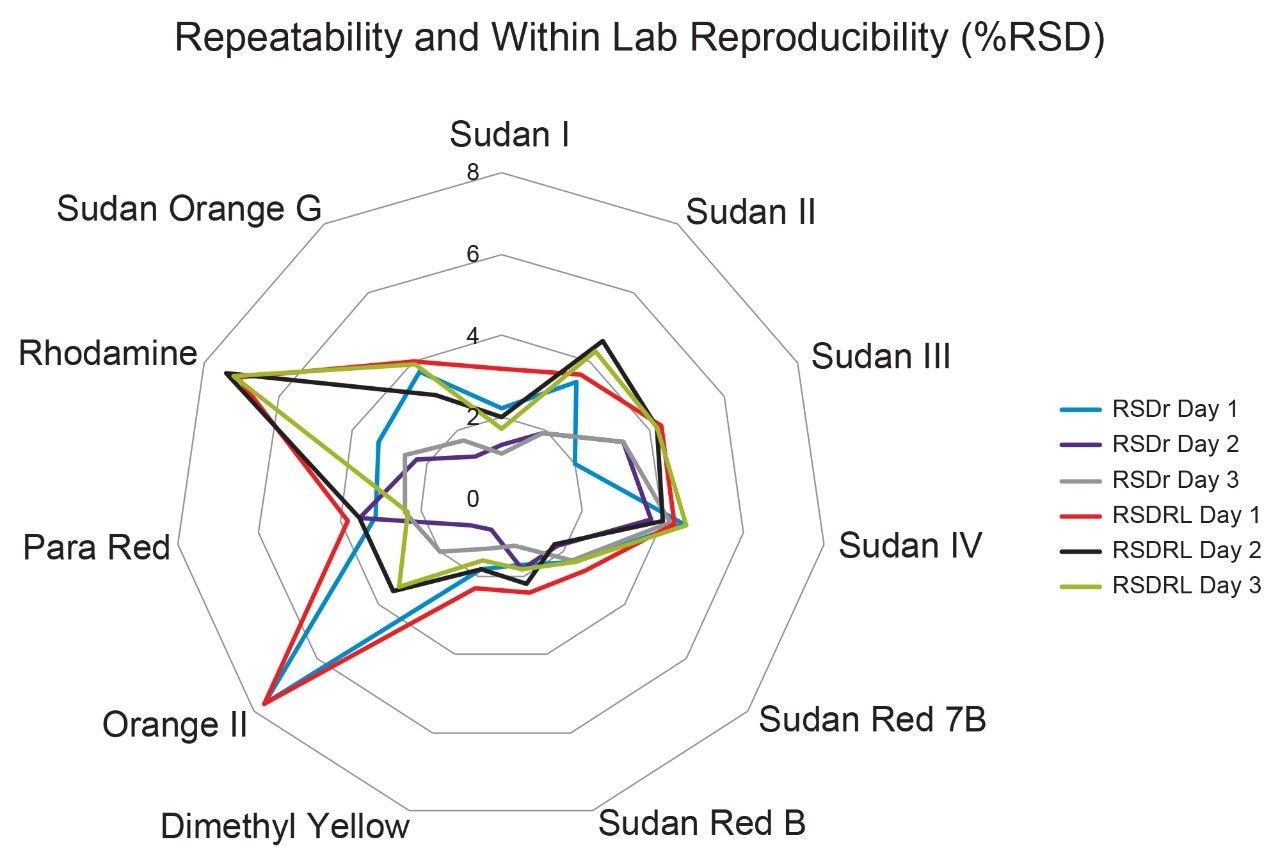
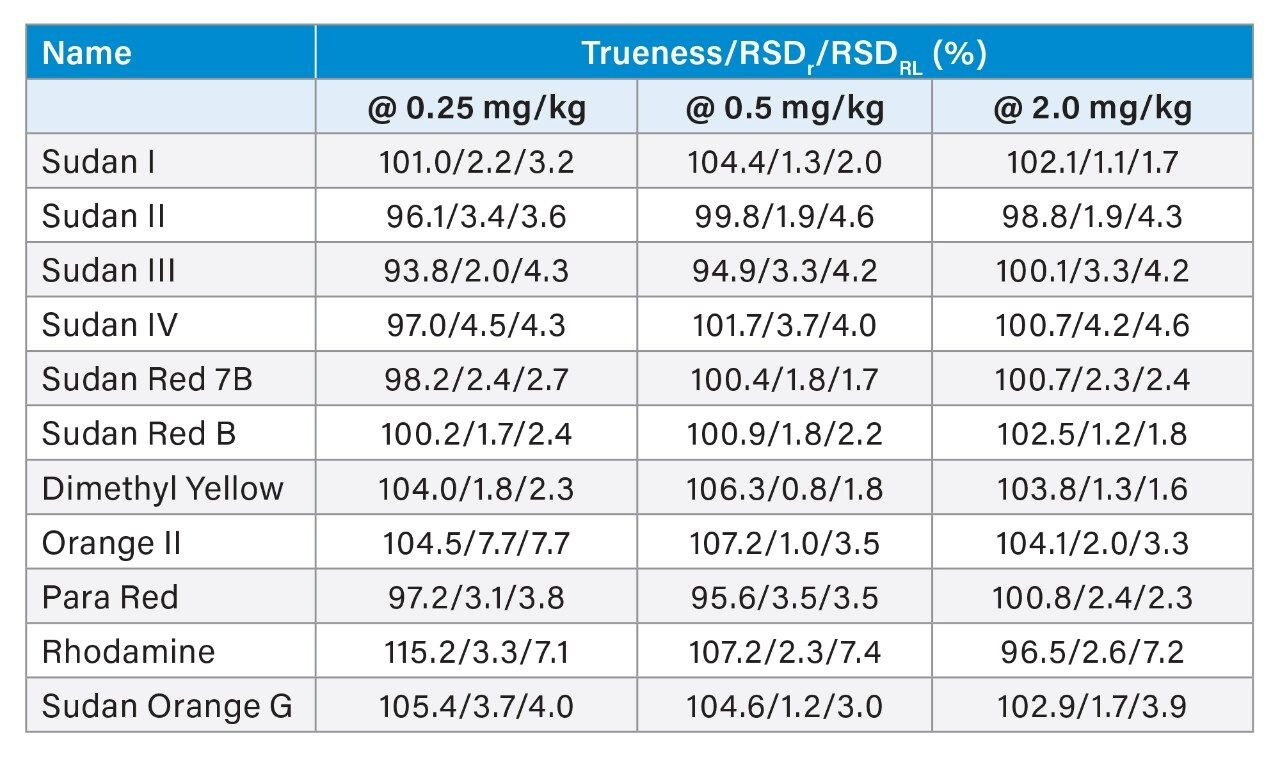
The combination of liquid chromatography with tandem mass spectrometry (LC-MS/MS) offers high sensitivity with outstanding selectivity and so has been extensively used in trace analysis of food. Here we demonstrate the application of a simple approach for the determination of illegal dyes in paprika using UPLC-MS/MS after extraction of 11 dyes with a strong organic solvent, using stable isotope analogues as internal standards, without any lengthy clean-up steps.