Data Processing - Bioinformatics
The acquired global expression datasets are processed using an algorithm that employs a maximum likelihood technique. This determines the exact mass, intensity, retention time and estimates precision for each eluting peptide species from an LC-MS dataset. Peak areas can then be normalized to an endogenous or exogenous internal standard. This provides the first output which can then be used for relative quantification, as exact mass retention time (EMRT) signatures are generated for every eluting species detected. Output of these EMRT signatures to programs such as Spotfire allows data visualization, or statistical treatment/clustering of the datasets to be performed.
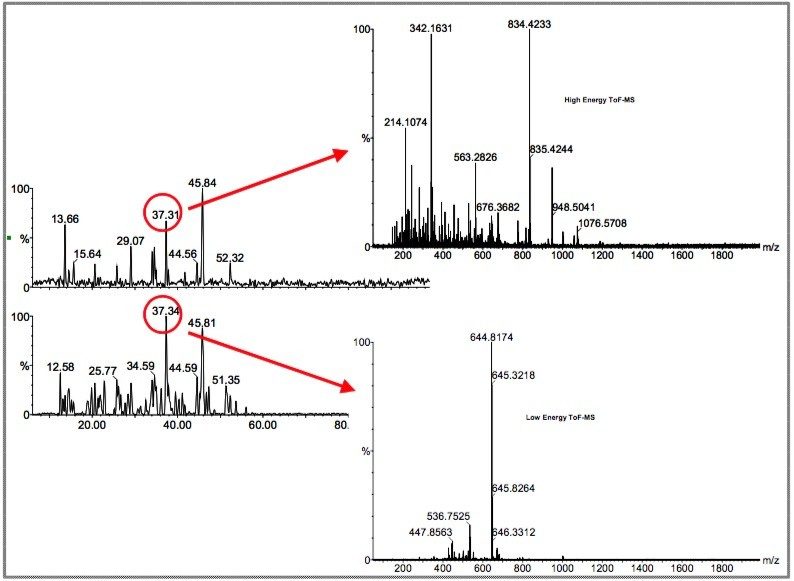
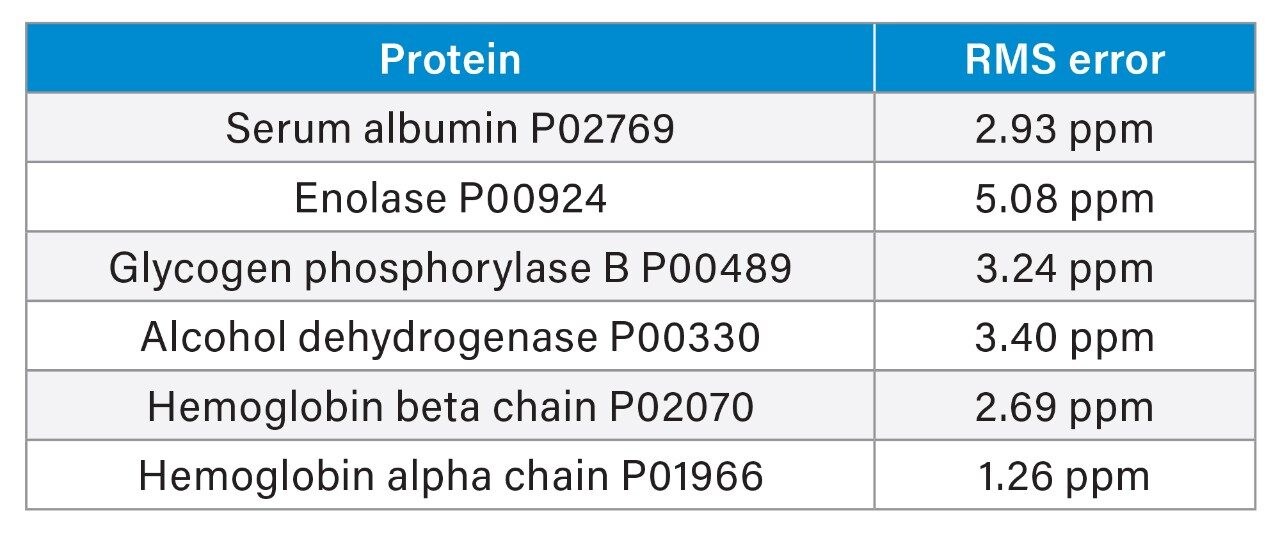
Further bioinformatic processing identifies the apex of each peptide ion identified in the low energy data function, and interrogates the MSE data function, acquired with an elevated collision energy, to reveal an associated set of exact mass fragment ions. By combining the exact masses of the peptide molecular ions and the associated fragment ions, a highly specific set of time resolved masses are generated that define that peptide. These time resolved sets of exact masses are searched against a databank using a proprietary peptide fragmentation model. Each matching protein in the databank is given a probability and confidence value.
The results are displayed in an interactive browser and list the matched protein sequences ranked by their probability. This display also allows visualization of the associated peptides that match to each protein sequence and display the relevant fragment ion data.
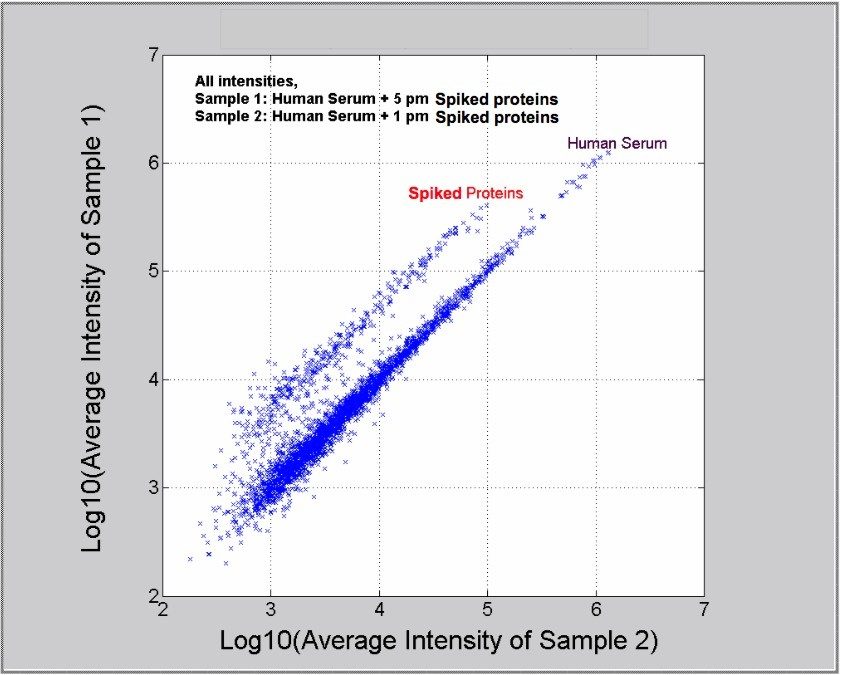
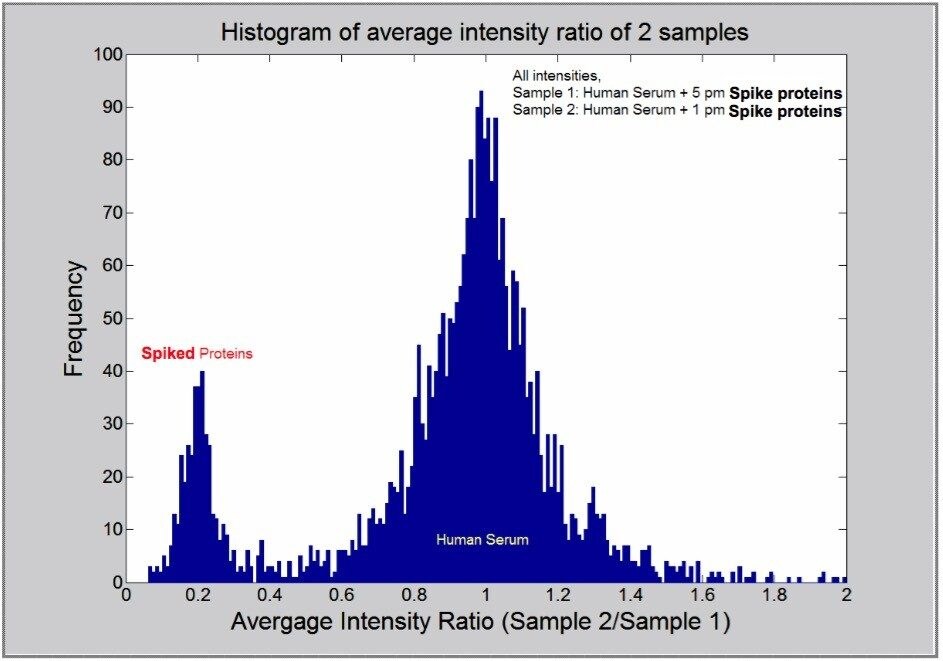
The expression ratio of proteins having a significant probability can then be determined across two or more sample sets, by comparing peak intensities of the relevant monoisotopic peptide masses. The probabilistic technique also provides a measure for the uncertainty of the ratios. Batch processing can be used to compare control with experimental samples.
These routines can be incorporated, as part of the Protein Expression System, in ProteinLynx Global SERVER v2.2.