The number of peptides in pre-clinical and clinical trials has seen steady growth over the past decade. Where development was previously hindered by factors such as efficient clearance rates and short half-lives, advances in formulation and alternative delivery systems have contributed to a resurgence.1,2 Peptides make up a unique class of pharmaceuticals not readily classified as small molecules or biologics. This becomes especially important in a regulatory framework. The United States Food and Drug Agency (FDA) has revised the definition of a biological product to include a protein (except any chemically synthesized peptide), where the term protein refers to a “defined sequence that is greater than 40 amino acids in size.”3 A chemically synthesized polypeptide must be made completely by chemical synthesis and be less than 100 amino acids in size.3 Effective March 2020, a protein will require submission of an application for a biological product. With this in mind, pursuing a synthetic peptide manufacturing strategy over recombinant strategies may become more appealing in an effort to reduce costs and deliver products to market more quickly.
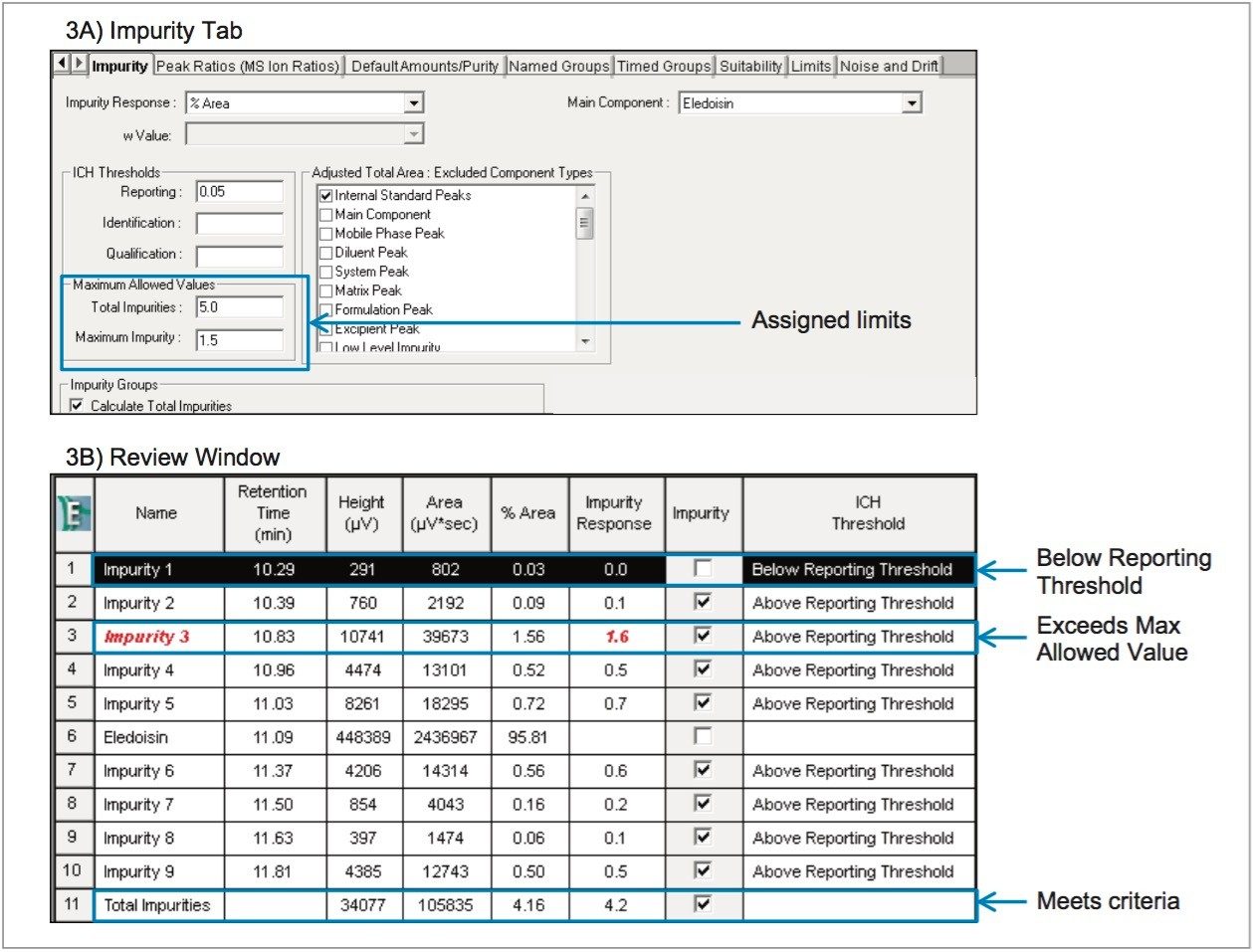
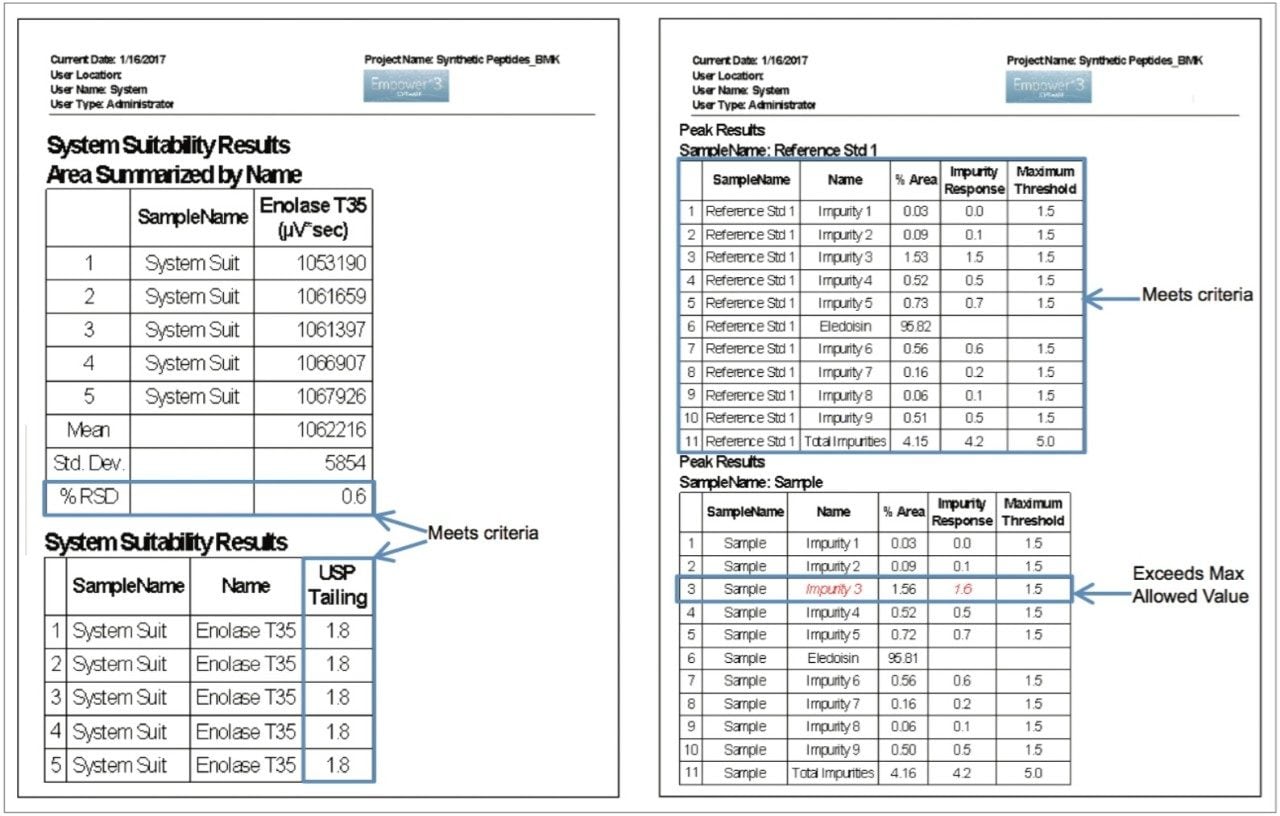
Analytical characterization and quality control of synthetic peptide products falls into one of four test categories: identification, assay, impurities, and specific tests. Impurities can result from the manufacturing process or from degradation during manufacturing or storage, and are typically determined by HPLC.4-6 Treatment of the HPLC data can suffer from a variety of user-induced pitfalls. Peak area integration, for example, can often be subjective without defined processing methods in place. Also, it is not uncommon to export results to external software for processing, which can introduce transcription errors or incorrect calculations, but can also create an added burden to maintain compliance. Informatics suites that support impurity profiling workflows and offer integrated data analysis options are highly desirable, both for eliminating user error and ensuring compliance.
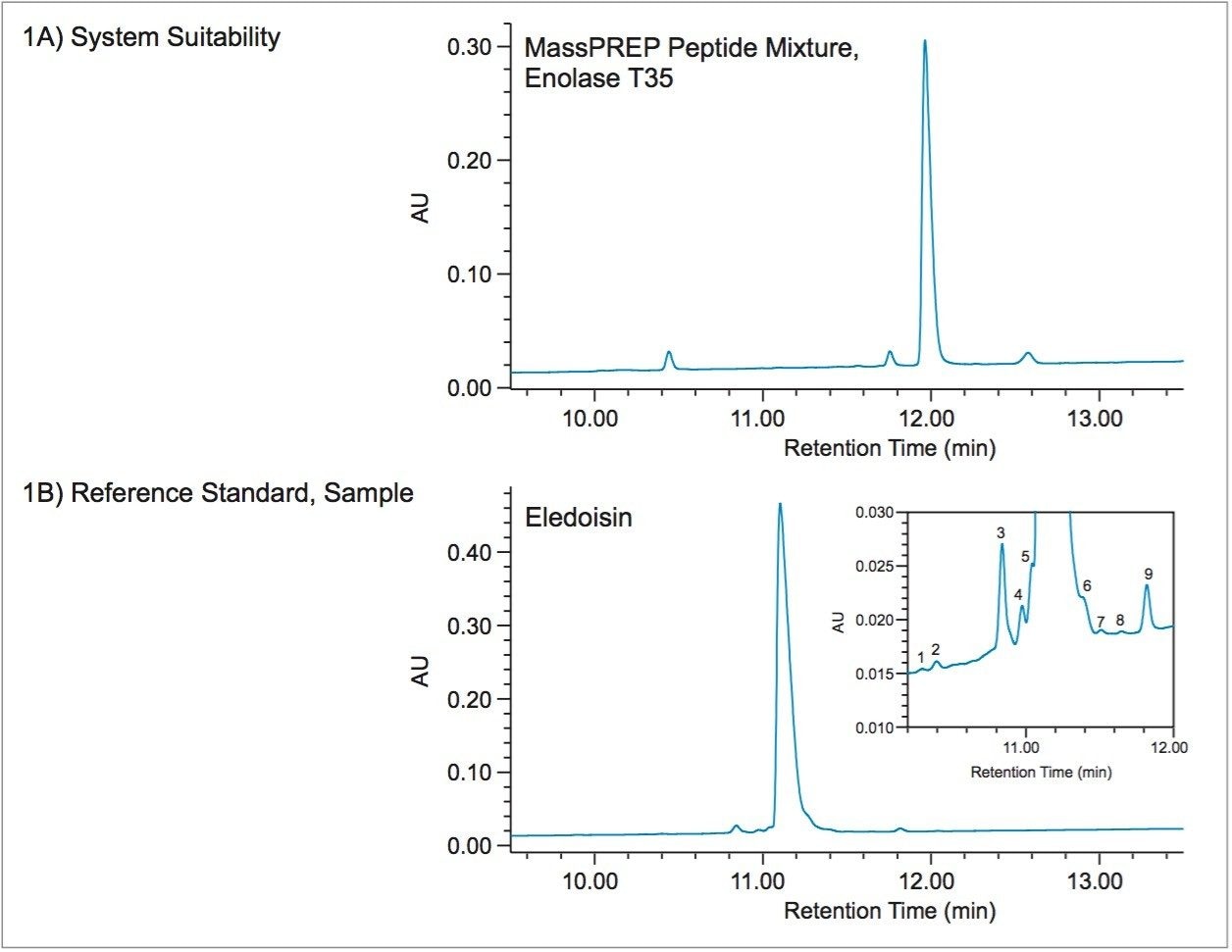
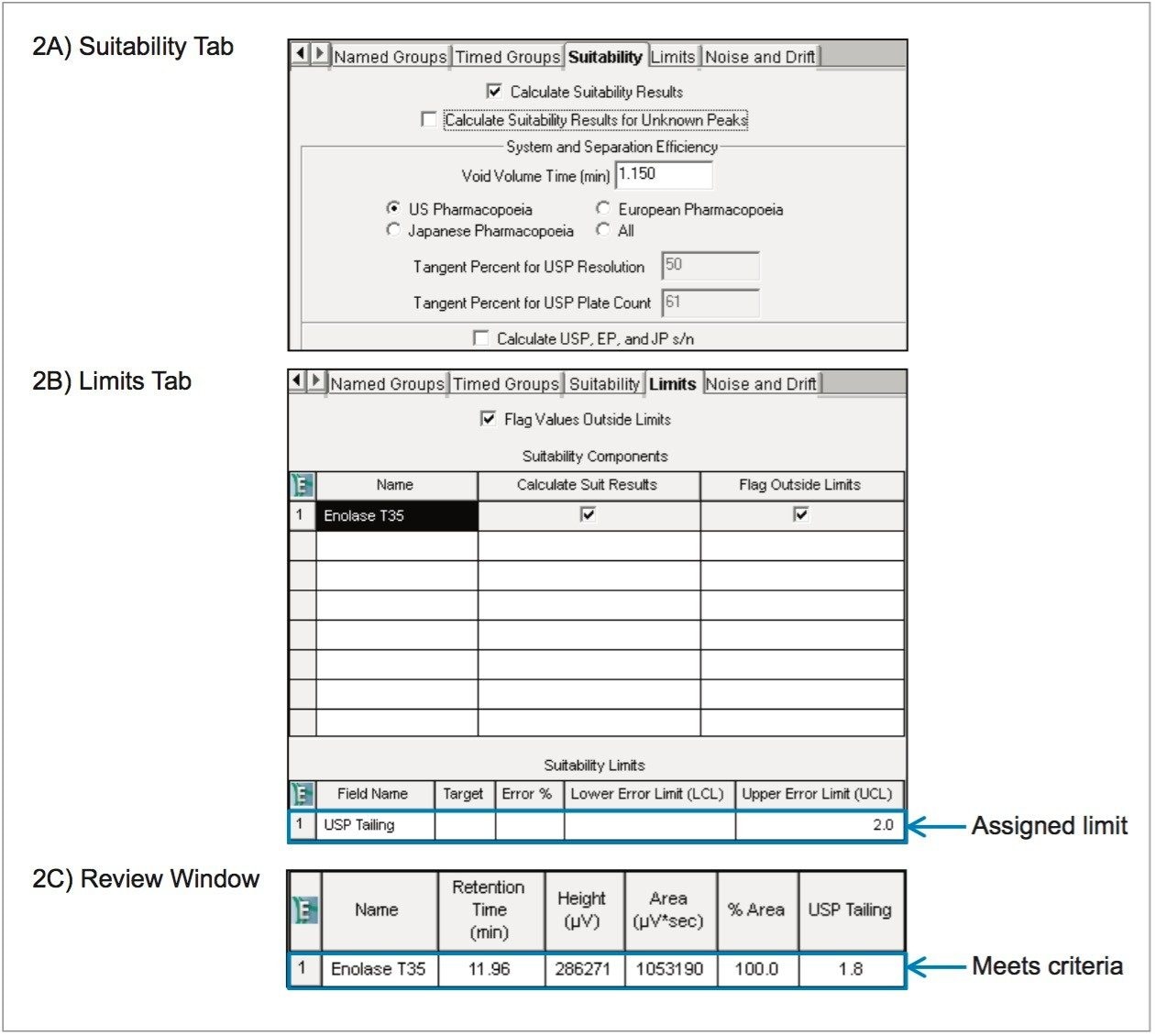
In this work, a UPLC-UV-based method is used for determining product purity in accordance with the International Council for Harmonisation (ICH) and United States Pharmacopeia (USP) functionalities built into Empower 3 Chromatography Data Software (CDS). The peptide used for this study was eledoisin, which is a biologically active peptide that acts as a vasodilator, and is thus used as a clinically relevant model system. Through establishing system suitability and impurity limits within the software, standards or samples not meeting acceptance criteria can be flagged and reported. By developing processing and reporting methods, future data can be handled in a relatively automated fashion.