U.S. Pharmacopeia (USP) compendial methods are routinely adopted by pharmaceutical companies during the development and quality control of drug substances and finished drug products. Often, these methods use mobile phases with non-volatile buffers, which are not suitable for mass spectrometry (MS). In some cases, mass spectrometry will enable quick and accurate identification of new or unknown components that may develop during the formulation process or routine testing. Incorrect identification or failure to identify these components may compromise the safety and efficacy of the pharmaceutical product.
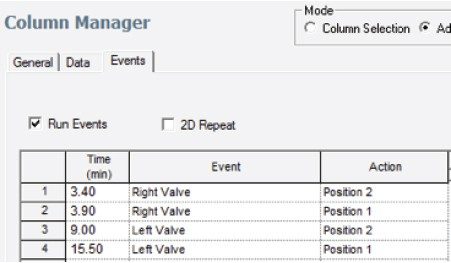
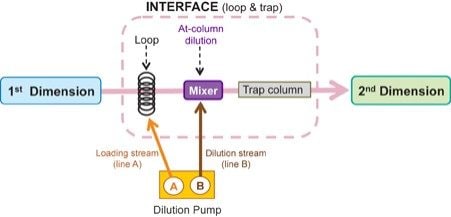
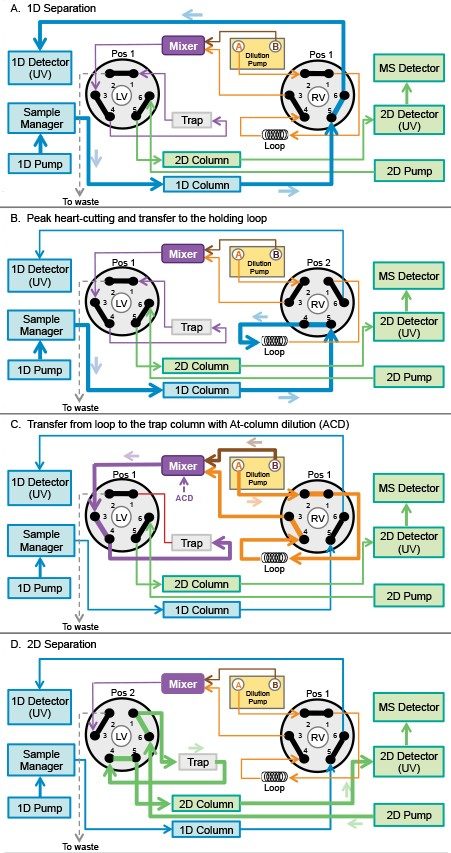
Changes to the USP monographs or registered methods to make them compatible with mass spectrometry will require a re-validation. In order to obtain mass spectral information for a sample tested using a non-MS compatible method, the sample must typically be isolated using a preparative LC system, purified, and reanalyzed under MS conditions.1 This process can be time consuming, and require additional instrumentation and development work. A multi-dimensional liquid chromatography with heart-cutting technique enables use of mass detection directly with the unmodified methods. With this system, peak of interest is heart-cut from the first dimension (1D) column eluted with non-MS compatible method and transferred into a holding loop. The heart-cut volume is then refocused onto a trap column and transferred to the second dimension (2D) separation column, which uses MS friendly conditions. This approach allows quick identification of unknown components by mass detection, eliminating the need to modify the method.
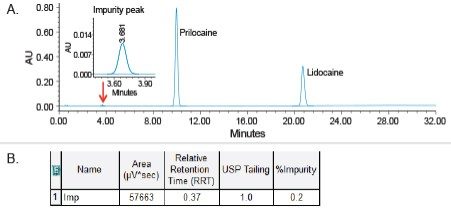
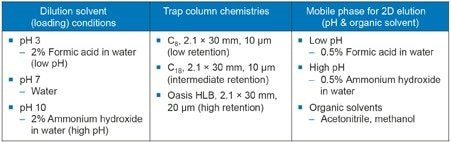
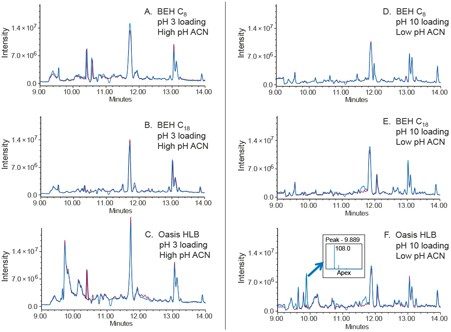
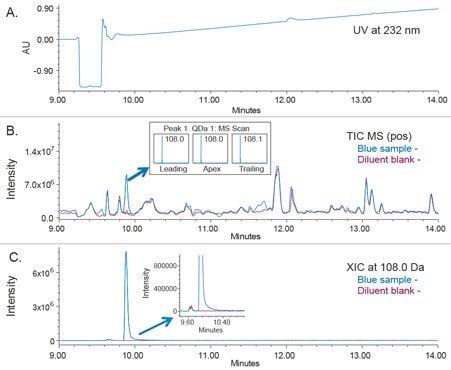
In this work, we demonstrate the capability of multi-dimensional liquid chromatography for the use of mass detection with an unmodified USP monograph for lidocaine and prilocaine cream2 to confirm identity of an impurity peak. An impurity peak observed in the assay preparation solution is heart-cut, trapped, and transferred to the second dimension for analysis by mass detection. The heart-cutting with At-column dilution approach for peak transfer and the development of conditions for maximum peak trapping (trapping column chemistries and dilution solvents) are discussed.