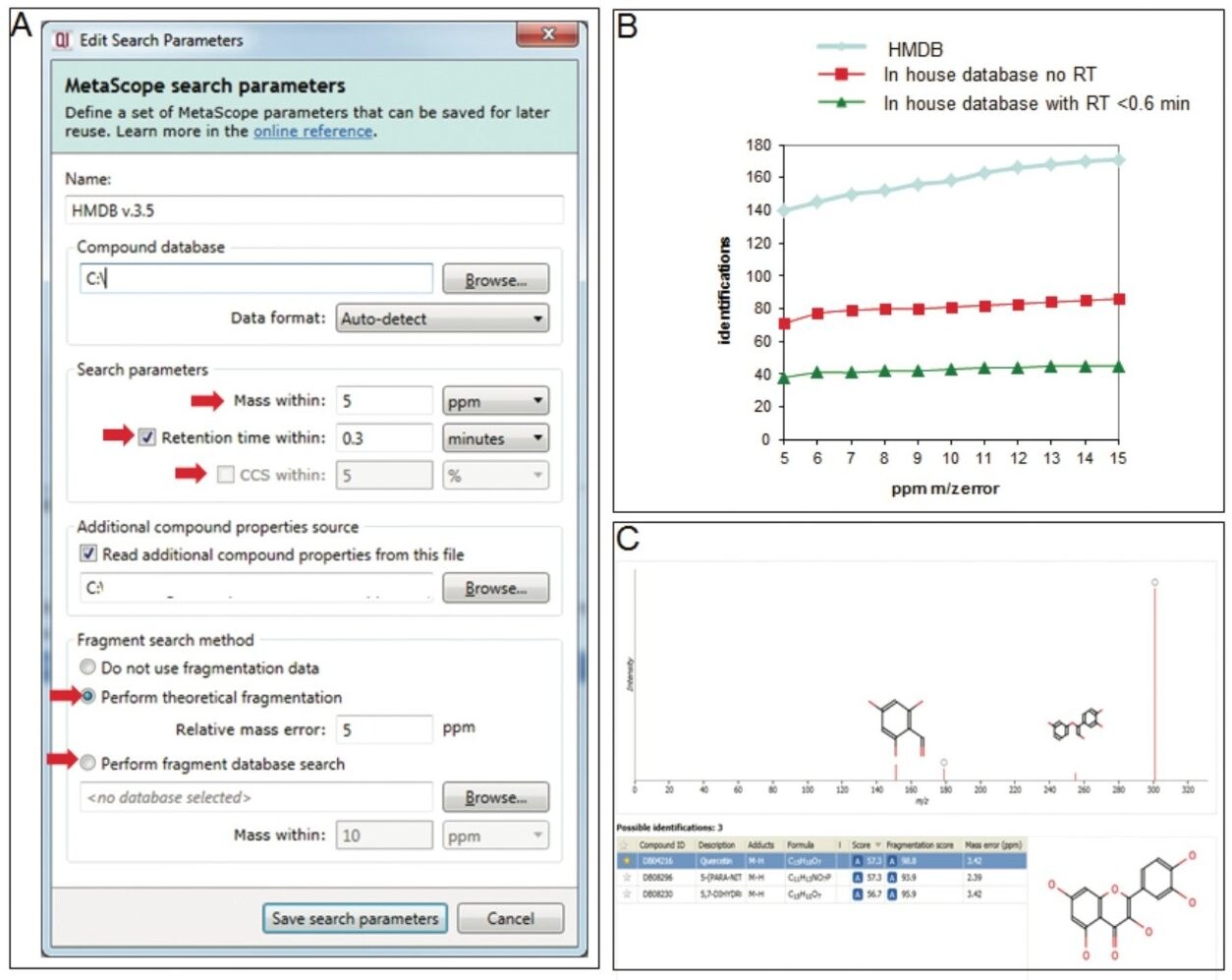
Metabolomics experiments offer a promising strategy for biomarker discovery. In a metabolomics workflow, however, the major bottleneck still remains metabolite identification. Currently, there are four levels of annotation for metabolite identification: 1) Confidently identified compound (two orthogonal properties based in authentic chemical standard analysis under the same condition); 2) Putative identified compounds (one or two orthogonal properties based in public database); 3) Putative identified compound class; and 4) Unknown compound.1 A typical database search that relies only on one property (i.e., accurate mass) usually leads to an extensive number of false positive and negative identifications. To increase the confidence of identification, a search engine should be able to use of in-house databases containing orthogonal molecular descriptors for each metabolite.2
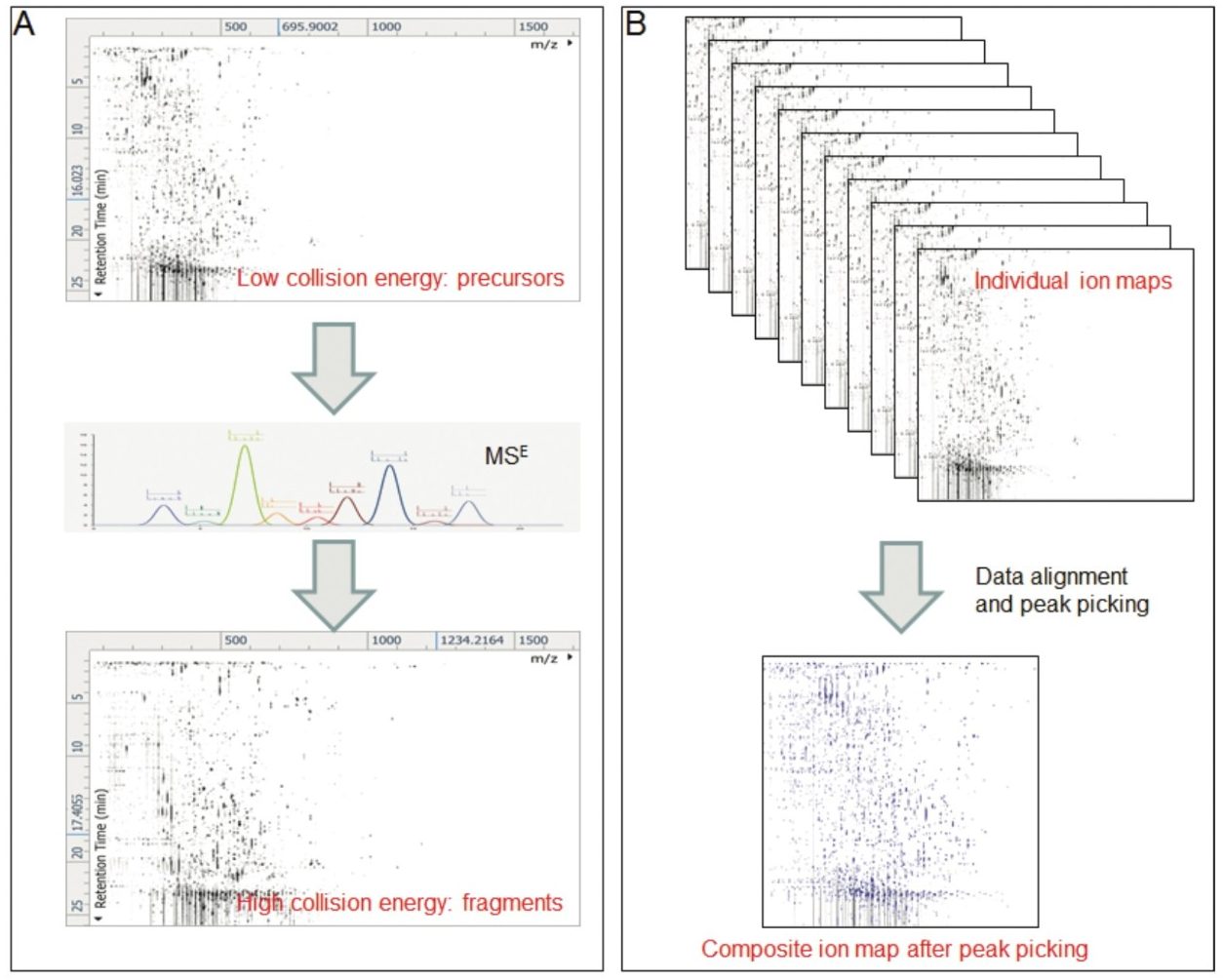
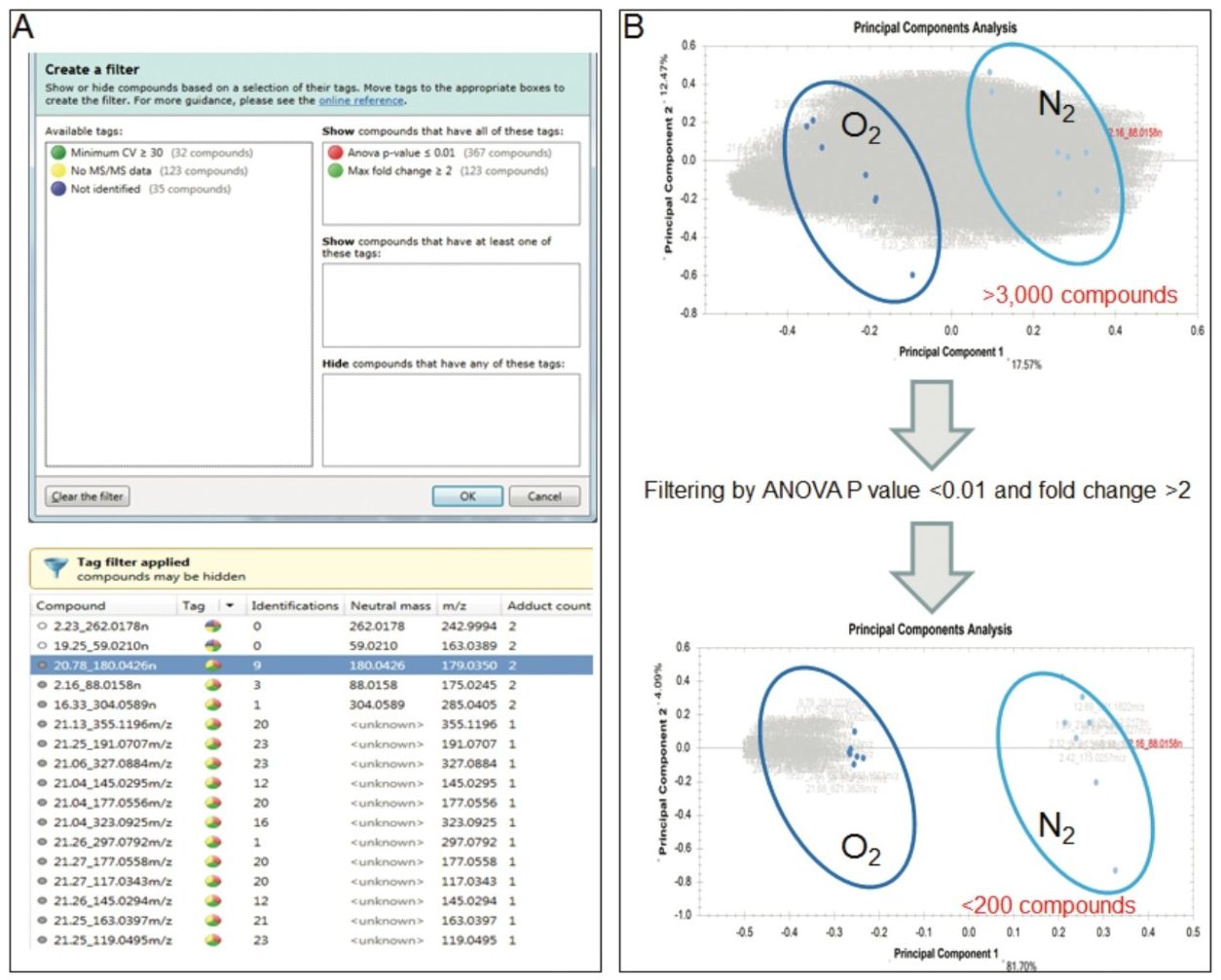
Progenesis QI Informatics is a novel software platform that is able to perform alignment, peak-picking, and mining of metabolomics data to quantify and then identify significant molecular alterations between groups of samples. The software uses a search engine (MetaScope) for metabolite identification, with user-definable search parameters to probe both in-house and publicly available databases. With an easy-to-use interface, the user can combine information for metabolite identification, including accurate mass, retention time, collision cross section, and theoretical and/or experimental fragment ions. These physiochemical properties can increase the confidence of metabolite identification while concurrently decreasing the number of false positives.
In this study, we show the Progenesis QI workflow for metabolite identification using, as an example, a study on the effect of different bottling conditions on the nutritional composition of Italian wines.