糖タンパク質分離に向けた HILIC テクノロジーの発展
HILIC は 1990 年代初頭に逆相クロマトグラフィーから採用した移動相を用いて高極性化合物を分離するための技術として始まりました9。 HILIC 分離メカニズムは、固定化された水和層を形成する極性の固定相によると主に考えられています9。 親水性分析種は固定化された水和層へ分配され、水素結合、双極子相互作用、イオン性相互作用により固定相と相互作用します。このように、親水性分析種は低極性の初期組成移動相条件下で HILIC 固定相に保持され、LC のグラジエントにより極性移動相の濃度を増加させていくことで、後ろに溶出されます9。
数多くの HILIC もしくは HILIC 様の固定相が過去 20 年位の間に開発されました。多くは非修飾シリカをベースとしたタイプで、ポリアルコールや両性イオンを結合したタイプなどの HILIC 固定相もあります。糖鎖の保持および選択性向上には、アミド結合相の使用が広まってきています。例えば、Waters Glycan カラムに用いられる ACQUITY UPLC Glycan BEH Amide 固定相は、高分離遊離糖鎖分析に向けて広く使用されています。
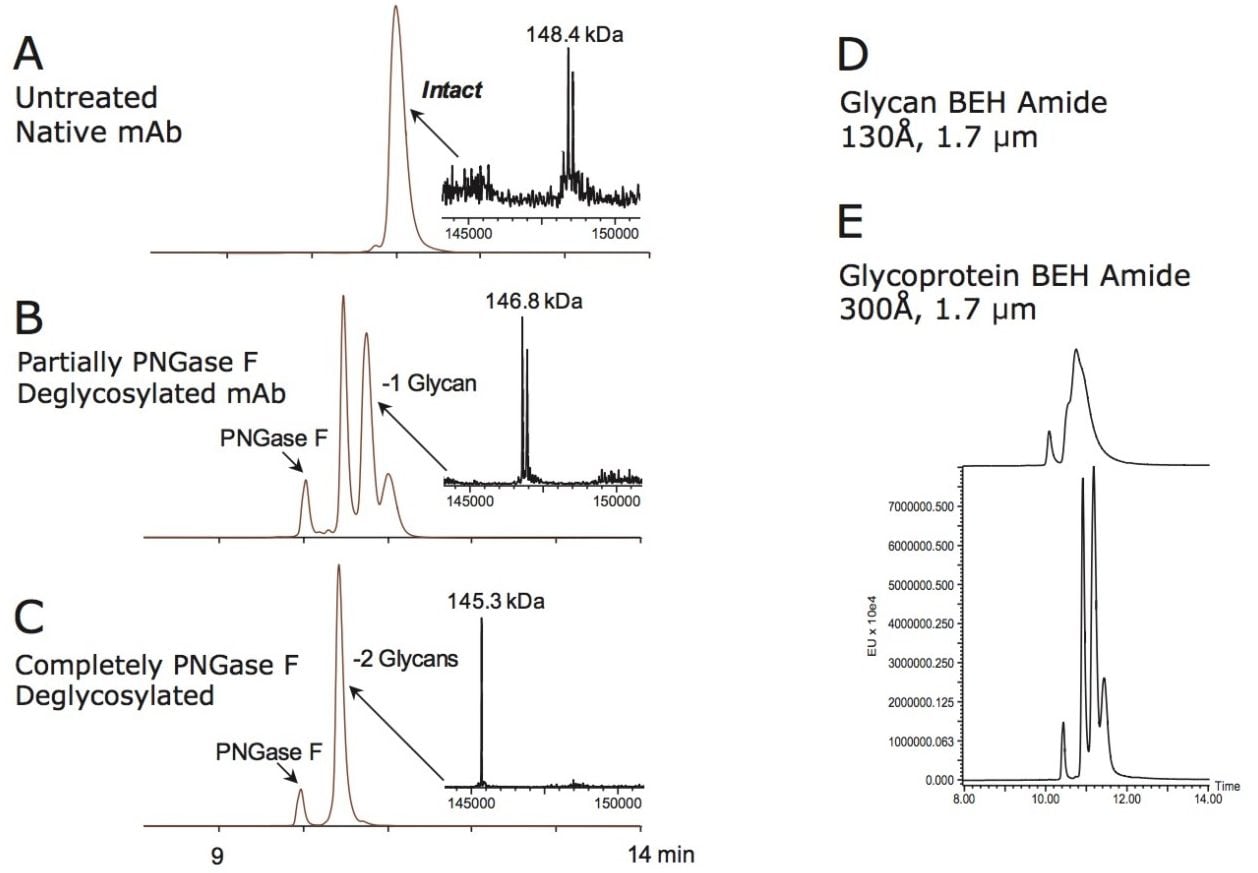
しかしながら、前述したように、HILIC はインタクトの高分子アプリケーションにおける広範な使用は見受けられません。高有機溶媒比率によるタンパク質の析出に関する懸念が、HILIC ベースのタンパク質分析法開発を試みるのを思いとどまらせる大きな理由です。この認識を乗り越えるために、我々は高分子分離を促進するために設計されたワイドポア有機シリカ(エチレン架橋型ハイブリッド:BEH)パーティクルベースのアミド結合固定相を新たに開発しました。この固定相は、タンパク質が充塡剤細孔内の大半にアクセスでき、分析種のサイズが固定相の平均細孔径に非常に近い場合(例、3 倍以内)に生じる拡散の制限による顕著なピークの拡散を起こさないような大きい平均細孔径です。
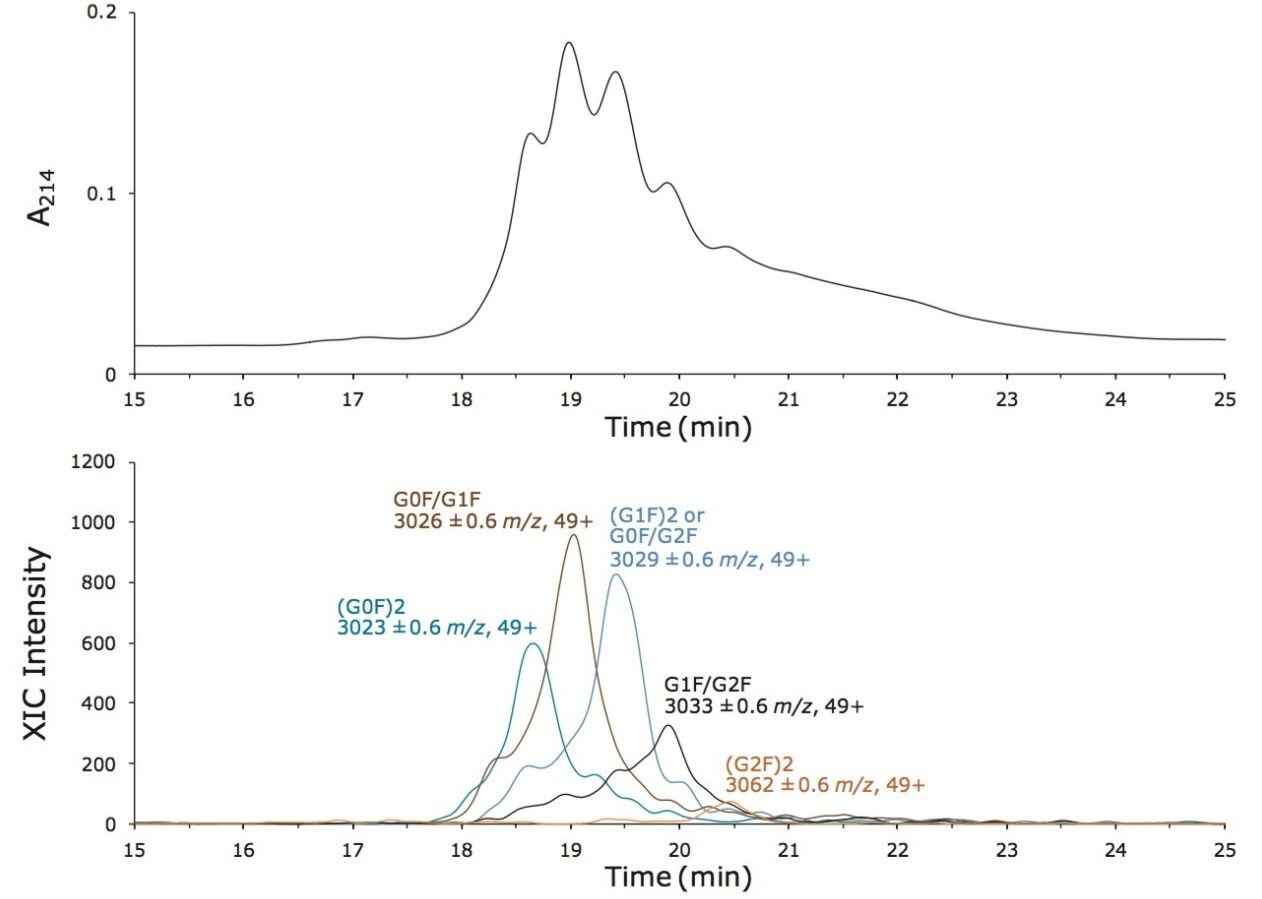
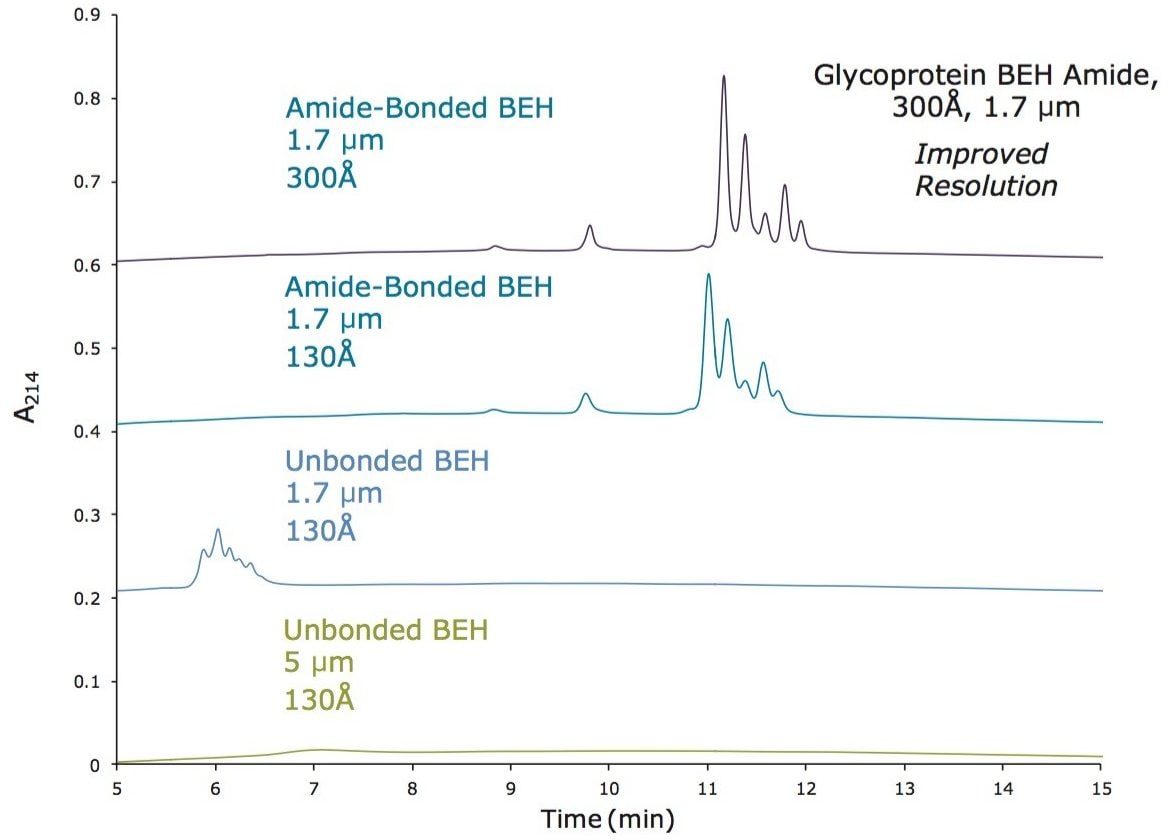
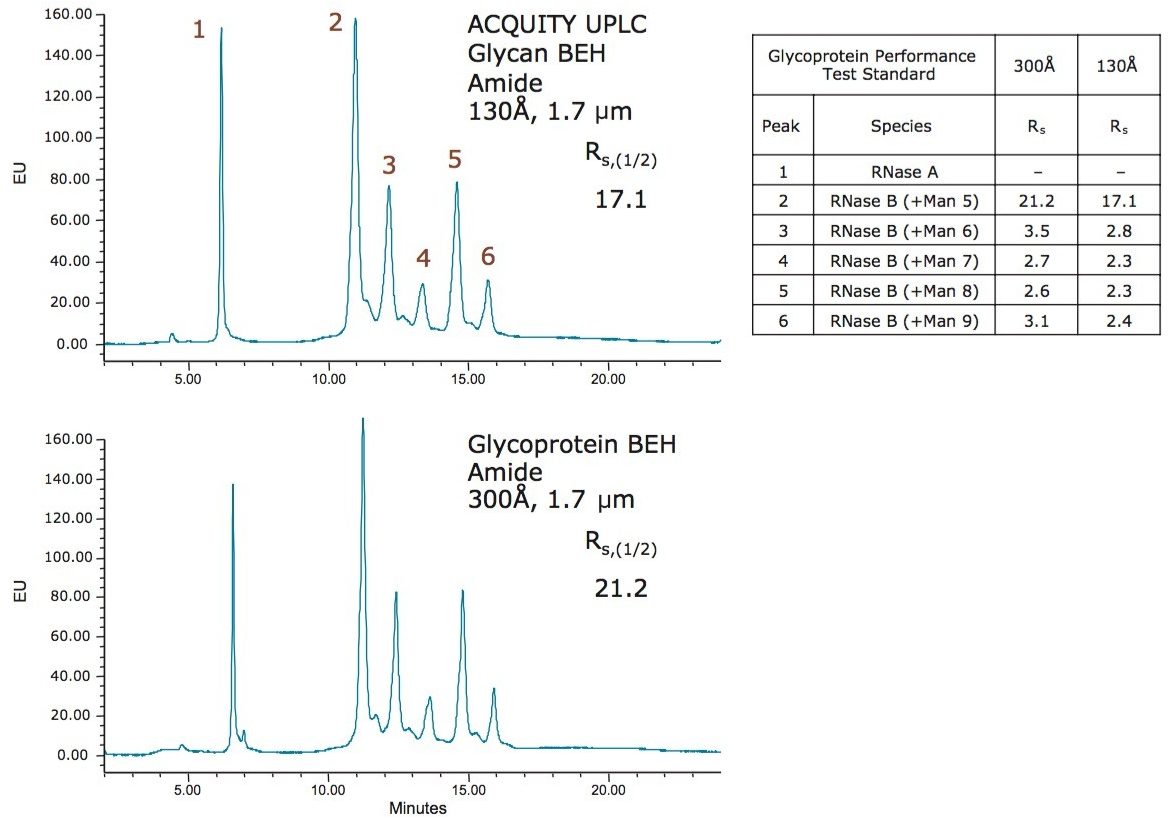
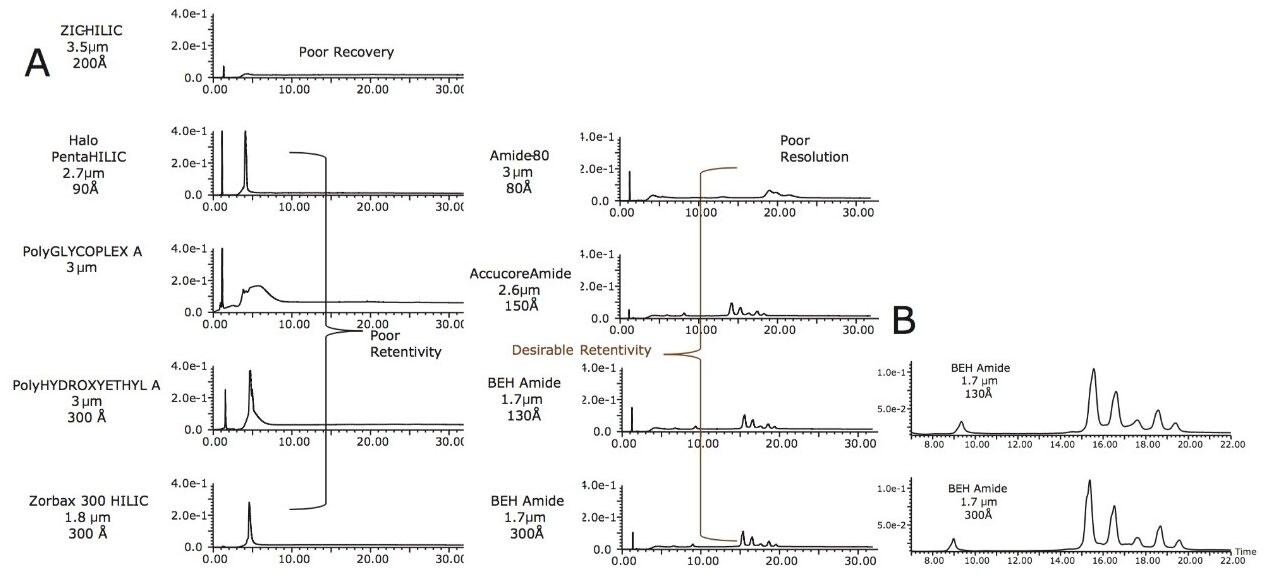
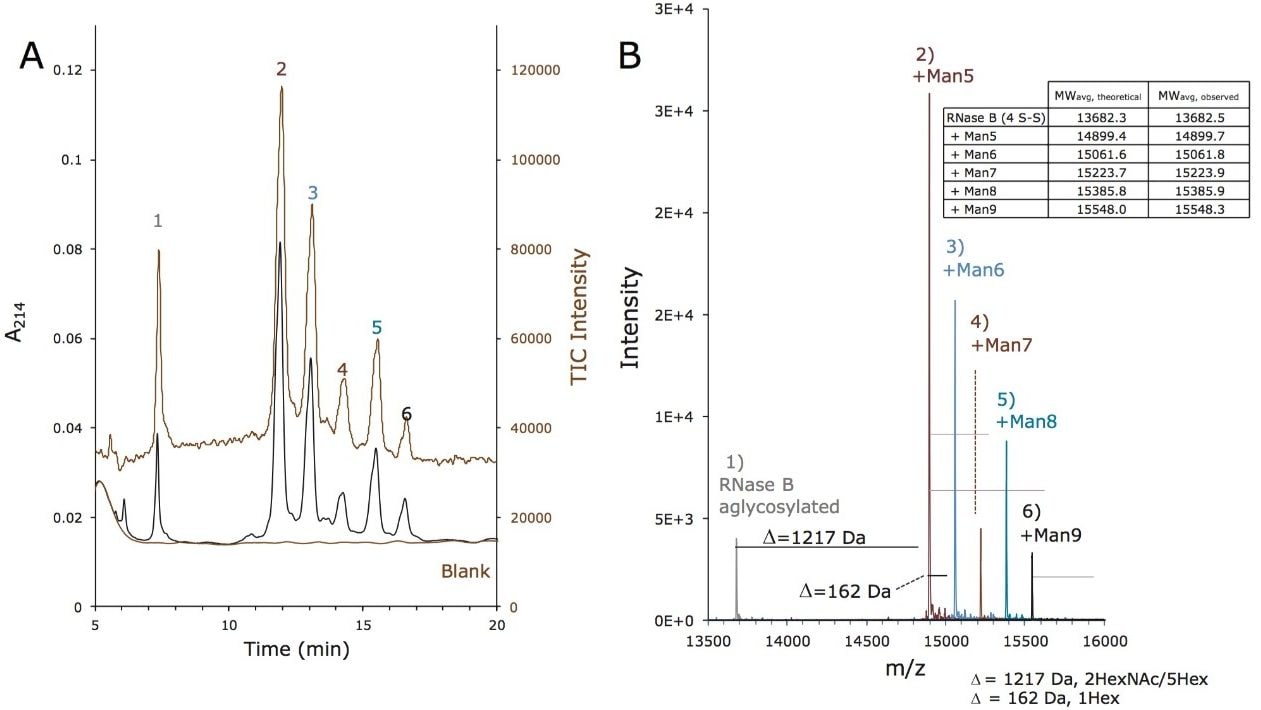
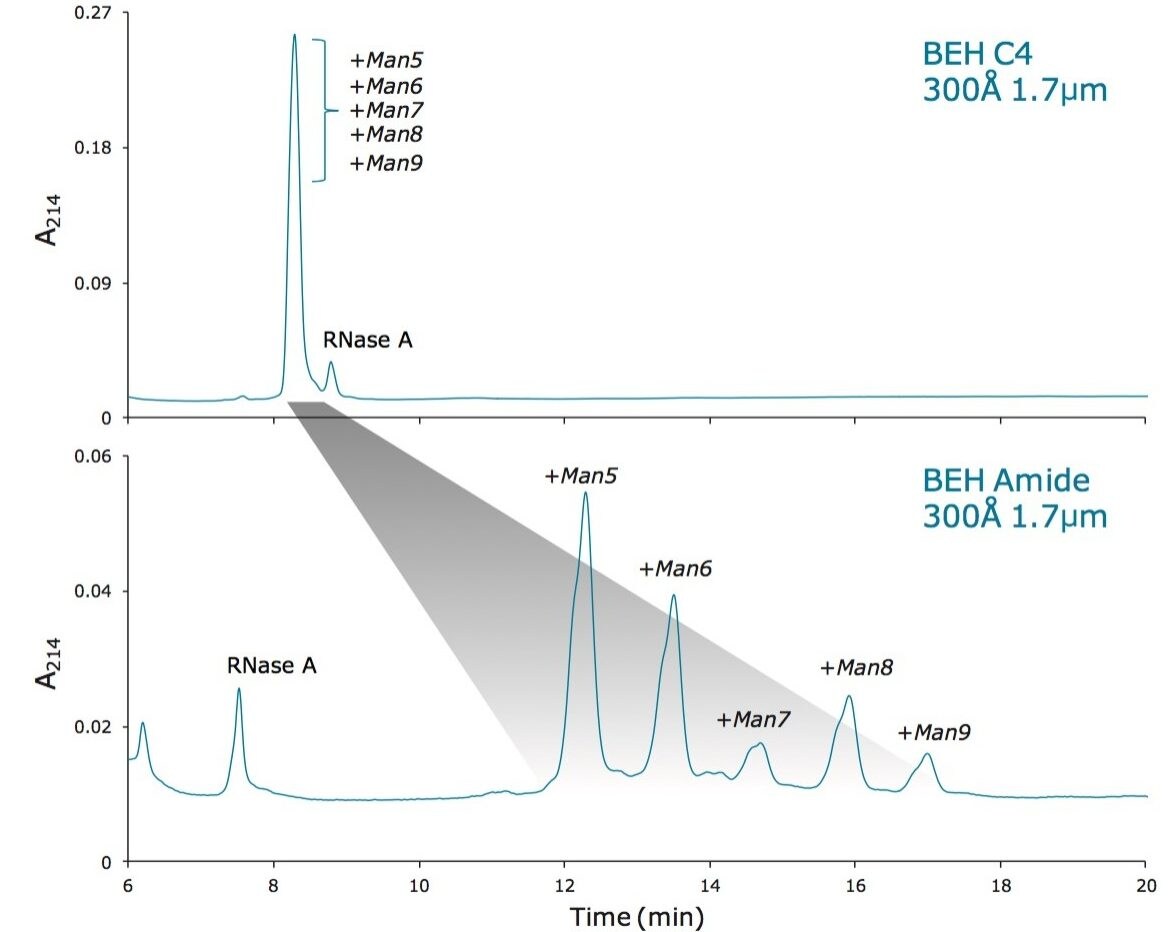
この新規固定相に至った HILIC テクノロジーの進歩は注目に値します。高分子 HILIC に向けたこの新たなテクノロジーは、いくつかのハイマンノース(Man5 – Man9)グライコフォームから成る 13 kDa のタンパク質である、ウシ ribonuclease B(RNase B)の分離に用いることができます。図 1 はいくつかの異なる固定相で分離した RNase B を示しています。下から上に行くにつれて、粒子径が 5 µm から 1.7 µm に、非修飾からアミド結合相に、スタンダードポアサイズ(130Å)からワイドポアサイズ(300Å)にと、より新規のクロマトグラフィーテクノロジーが採用され、より良好な RNase B の分離が達成されています。RNase B のグライコフォームが最も良好に分離したのは、BEH Amide、300Å、1.7 µm パーティクルです。ワイドポア固定相の使用が、最適な分離の達成に重要な役割を果たしています。ウシ RNase B、そのグライコフォームと脱グリコシル化したアイソフォーム(RNase A)を含む新規開発試験混合物(Glycoprotein Performance Test Standard)を用いて示した評価結果を図 2 に示しました。本スタンダードを用いた分離例では、ワイドポア(300Å)BEH Amide固定相、スタンダードポア(130Å)BEH Amide 固定相両方にフォーカスグラジエントが使用されています。ワイドポアアミドカラムでは、分離全体に渡って大きく向上が見られたことに加え、脱グリコシル化した RNase A アイソフォームと RNase B の Man5 グライコフォームの分離度が顕著に(24%)向上しました。
![IgG の HILIC 分離におけるカラム背圧の影響。トラスツズマブ(1 µg)を Glycoprotein BEH Amide、300Å、1.7 µm、2.1×150mm カラムと流路制限有りもしくは無しで分離[PDB:1IGT]。](/content/dam/waters/ja/app-notes/2015/720005380/720005380en-f7.jpg.82.resize/img.jpg)