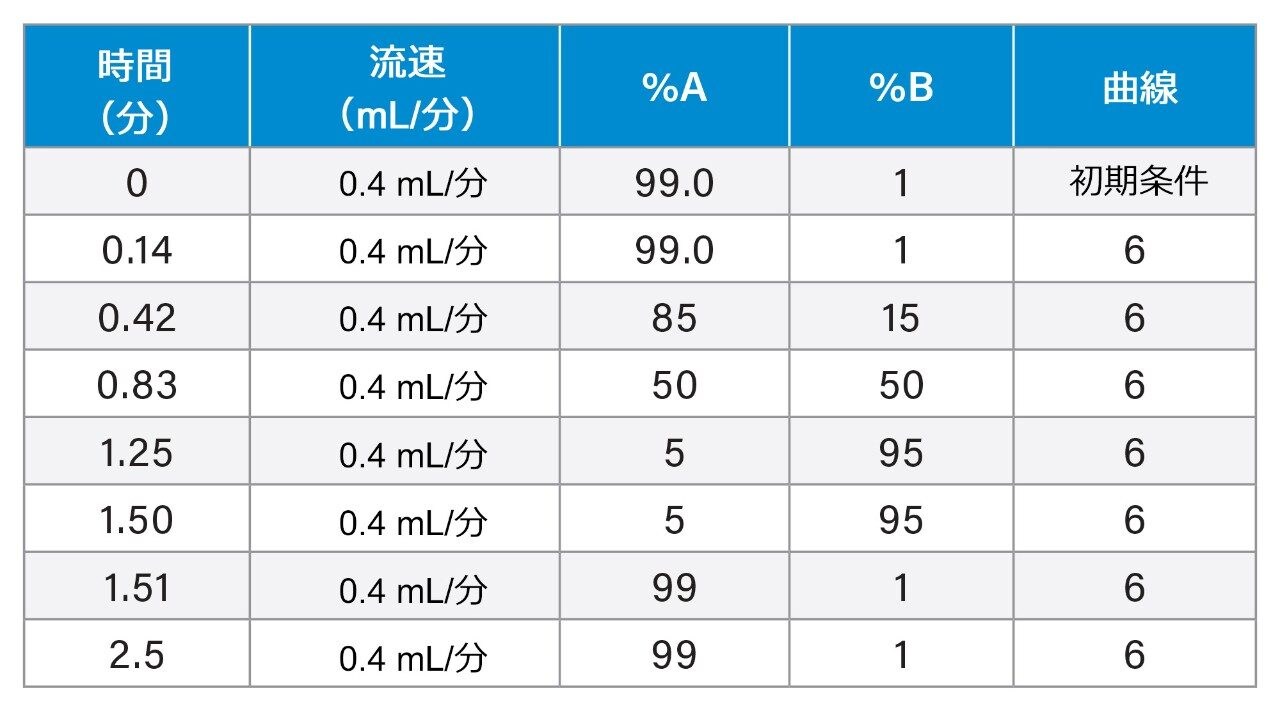
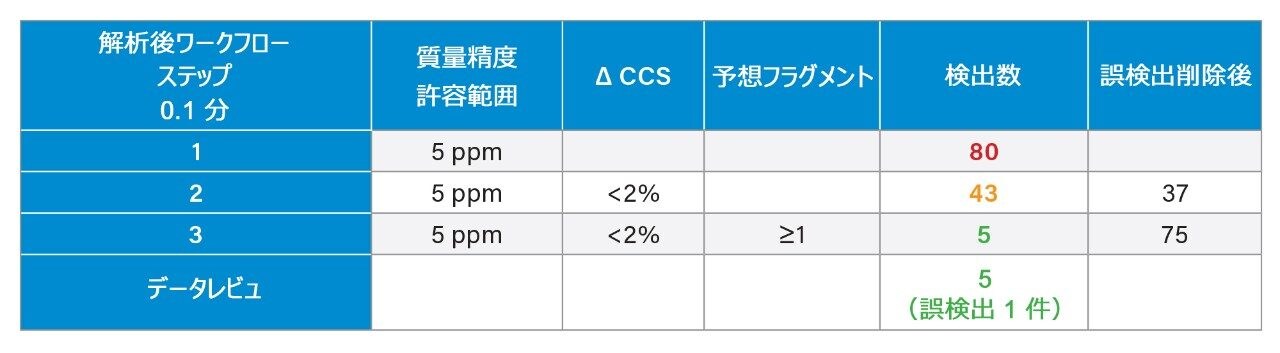
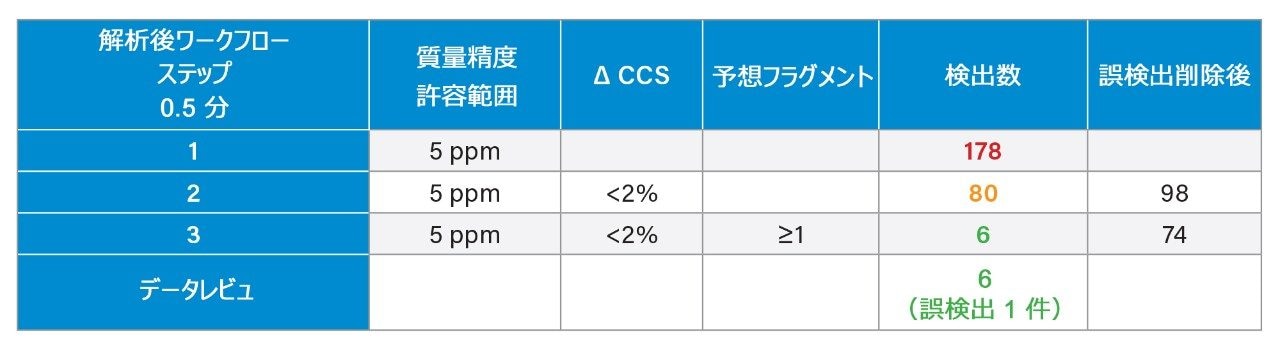
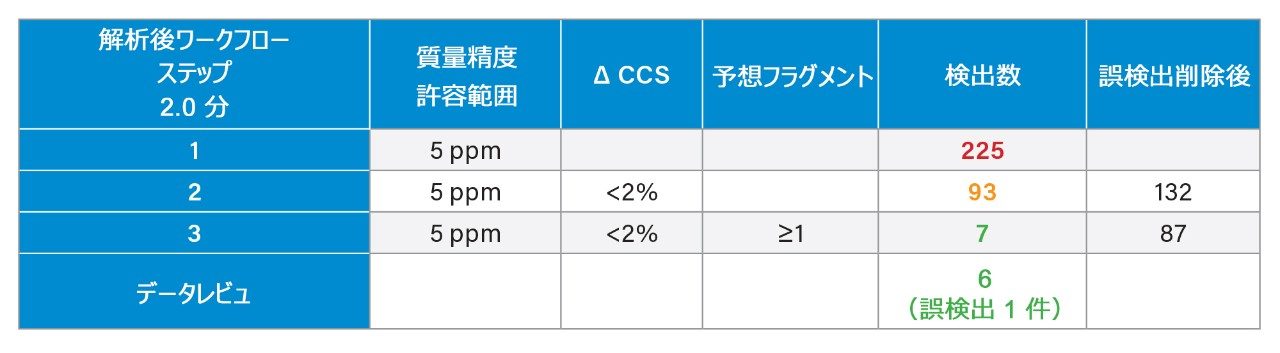
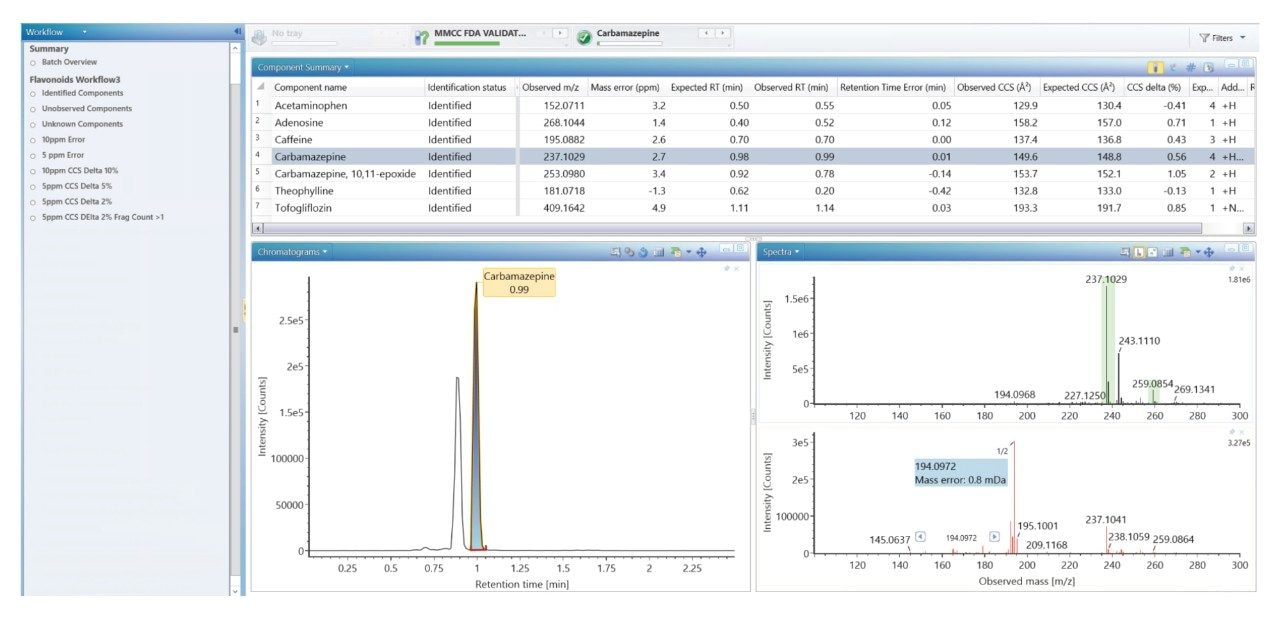
質量分析ライブラリーの作成に関する以前に説明されている戦略2 を用いて、FDA が承認した一連の低分子医薬品を、高速グラジエントマイクロボア超高速液体クロマトグラフィーイオンモビリティー質量分析(RGM-UPLC-IM-MS)を使用して特性解析しました。作成したライブラリーを使用することで、四重極ベース、ToF ベース、または IM ベースの分析が容易になります。採用した戦略により、分析種の保持時間(tr)、プリカーサーイオン、プロダクトイオン、衝突断面積(CCS)のデータが得られます。質量分析ライブラリーは、合計サイクル時間 12 分(2.1 mm × 100 mm のカラムを使用、溶離液流速 0.5 mL/分)の従来の UPLC メソッドを使用して事前に作成されました。ここでは、高速マイクロボア代謝プロファイリング UPLC-MS メソッドを使用して、同等の高速グラジエントマイクロボア UPLC-IM-MS ライブラリーを生成しました。この場合、1 mm × 50 mm のカラムで、溶離液流速 0.4 mL/分 を採用し、これらの条件によって合計分析サイクル時間が 2.5 分に短縮されました。従来のメソッドおよび高速グラジエント UPLC-MS メソッドで、ピークキャパシティはそれぞれ 150 および 50 でした3。
四重極飛行時間型(Q-ToF)質量分析部などの高分解能質量分析計(HRMS)は、臨床、法医中毒学、代謝物同定の各研究領域のスクリーニングツールとして、より広く使用されるようになりました4,5。ノンターゲット「フルスキャン」データ取り込みでは、1 回の分析で何千もの検出を行うことができ、その後、過去に遡ってターゲットデータ分析が行われます6 。多くの研究分野(農薬、マイコトキシン、天然植物毒素の試験など)では、サンプルスループットの向上、時間効率の向上、コスト削減が求められており、その結果、マルチクラス化合物分析に移行しました7。サンプルの複雑さを考えると、遺伝子型、環境、ライフスタイルの間の相互作用を分子レベルで理解するための代謝表現型解析にも、このような目標が存在します。これらの調査は、何千もの前臨床代謝/毒性、臨床、疫学的サンプルで構成される場合があります3。
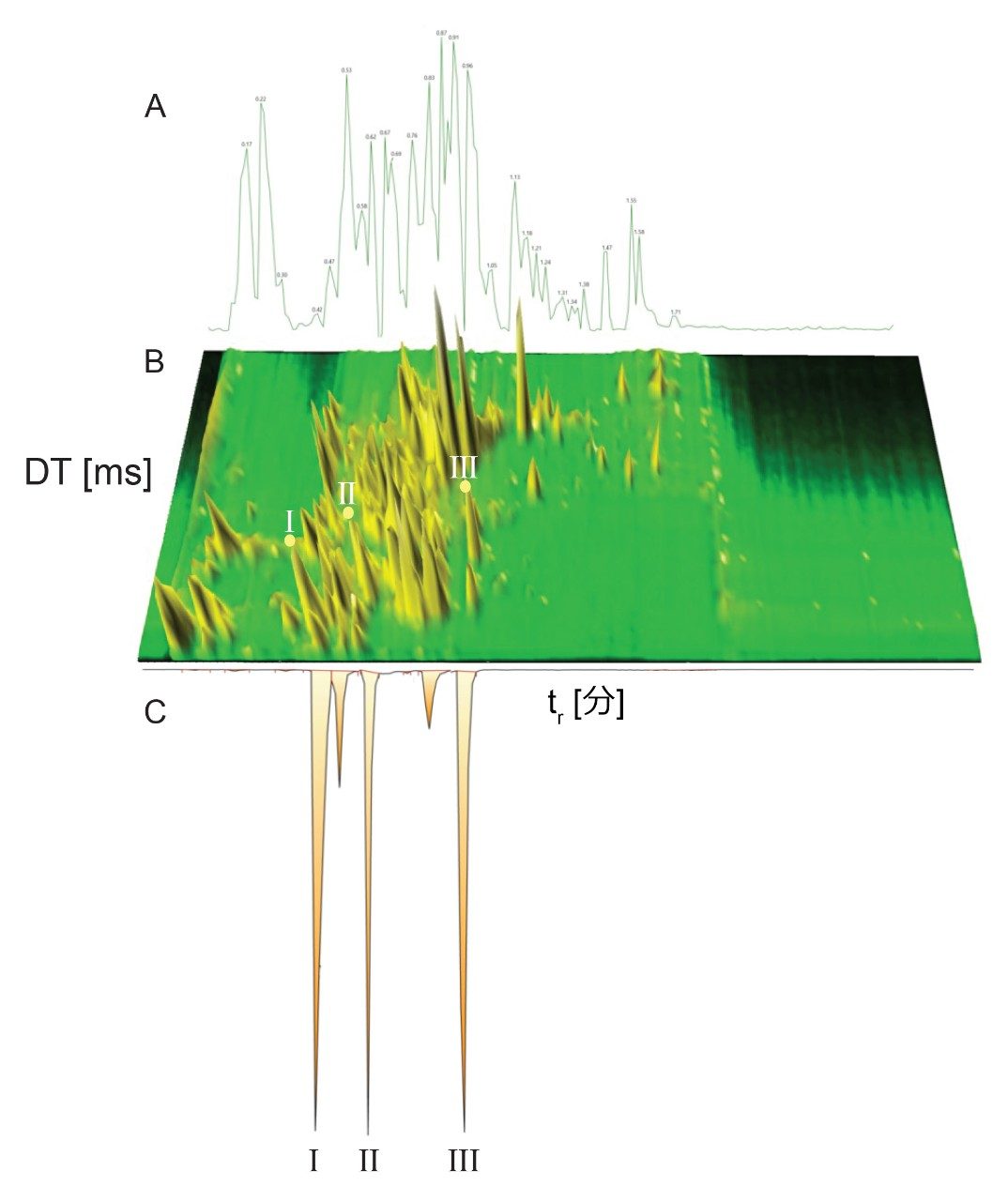
Dwivedi らは、LC、MS、および IM の直交性について議論し、MS および IM の分解能に応じて、ピークキャパシティが 2 ~ 10 倍に増加することを示しています8。RGM-UPLC メソッドとイオンモビリティー質量分析を組み合わせることで、ピークキャパシティ、特異性、分析のフレキシビリティが向上すると同時に、戦略の時間効率も維持され、サンプルスループットが 5 倍向上します9,10。UPLC-IM は、UPLC(中性分子種の分離)と組み合わせたイオンモビリティー分離(MS 分析前の気相分離)で構成されます11,12。UPLC(秒)、IMS(ミリ秒)、飛行時間型 MS(マイクロ秒)の所要時間は、複雑なサンプルのハイスループット分析の要件に適合しています。化合物のイオンモビリティー分離は、質量分析部の前のガスを充塡した進行波イオンモビリティー(TWIM)RF イオンガイド内で、気相イオンが分離されることによって行われます。モビリティー分離は、比較的弱い電界を使用して、イオンのパケットを不活性バッファーガス(窒素)中を通過させることによって行われます。イオンとバッファーガスの間の衝突回数により、ドリフト時間の差が生じます。結果として得られる分離は、RF イオンガイドに沿った DC パルスの繰り返し印加に基づきます。イオンは周期的にパルスまたは波に追い越され、モビリティーが小さい分子種はモビリティーが大きい分子種より追い越される頻度が高くなります。このため、デバイスを渡る時間はモビリティーに依存し、イオン質量、電荷、形状などの要因に支配されます。イオンモビリティーにより、補完的な同定規準である CCS(衝突断面積)に加えて、LC(疎水性)および MS(m/z)での分離に、別の次元の分離が追加されます。CCS は同定の特異性を高め、代謝物同定、農薬の分析、マイコトキシンのスクリーニング、薬用植物の分析、食品添加物の分析など幅広いアプリケーションにわたってその有用性が実証されています10,13-17。
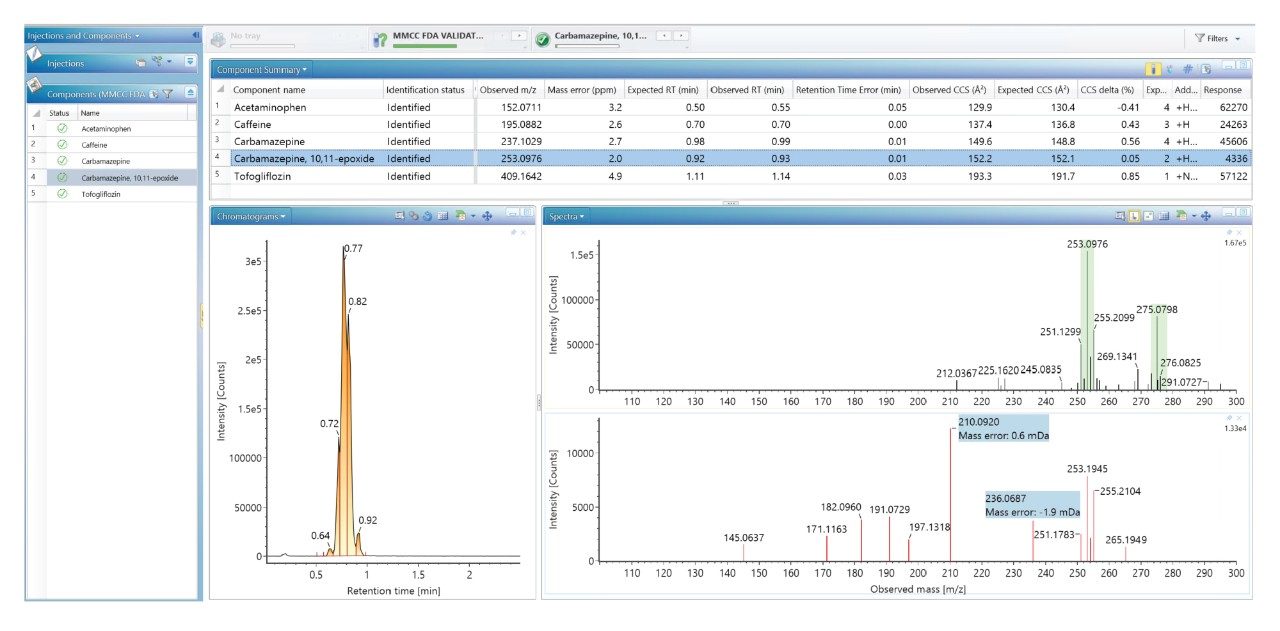
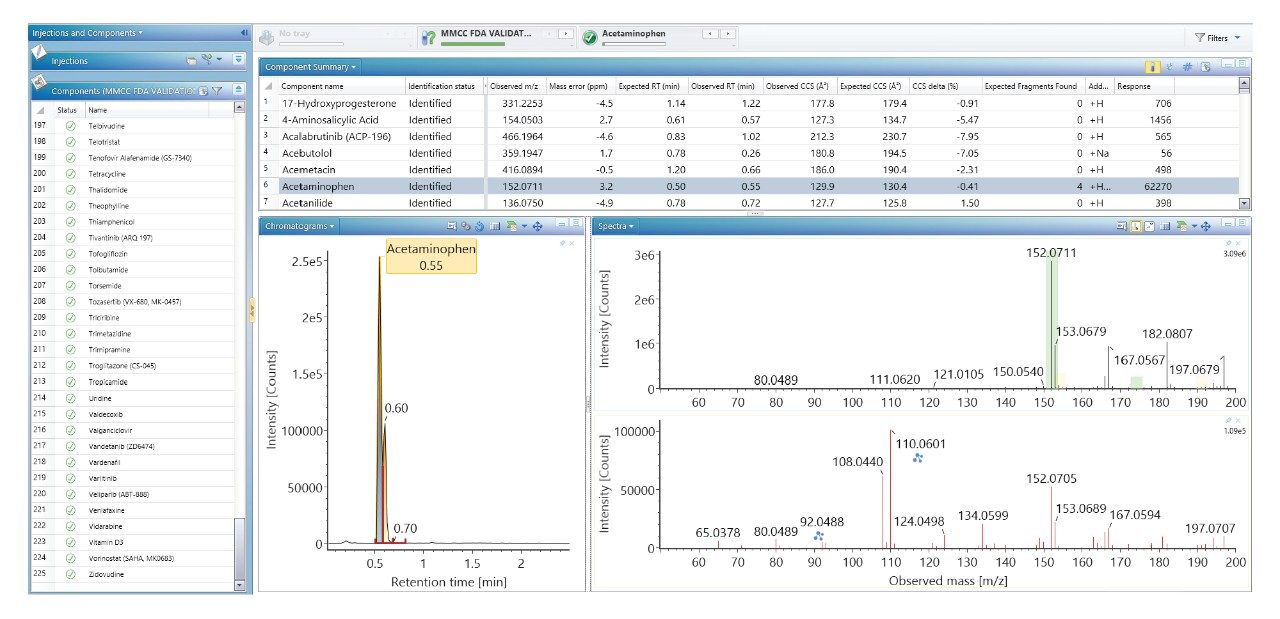
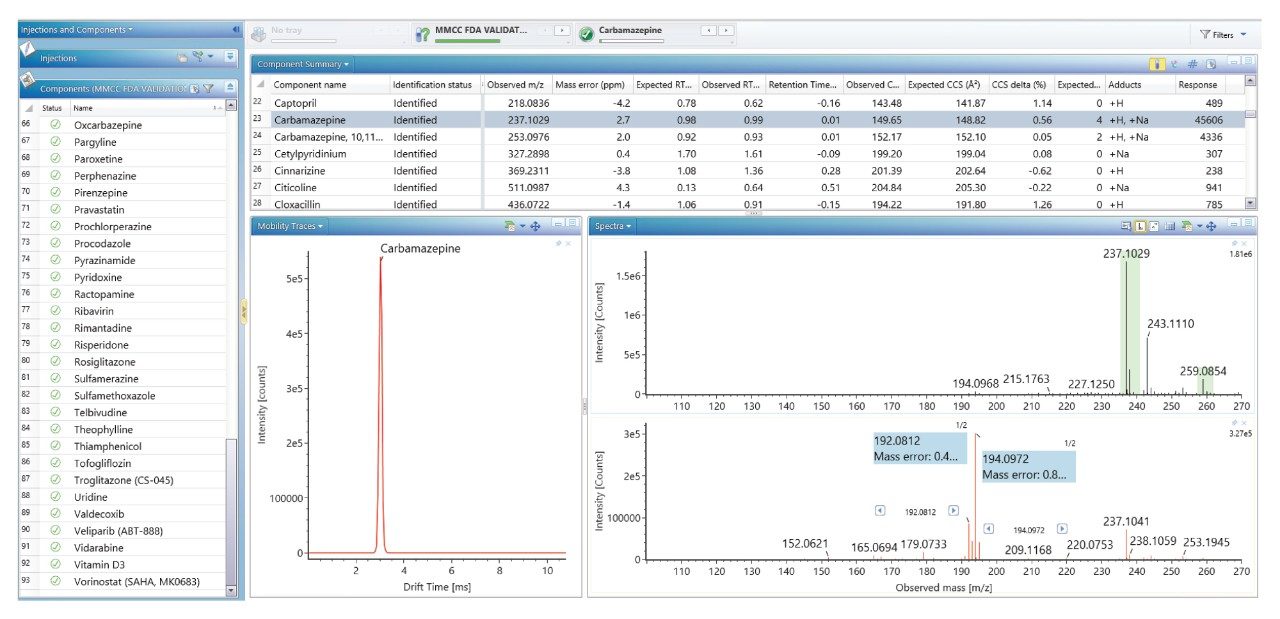
当社では、FDA が承認した一連の低分子医薬品のデータのライブラリーを使用して、健常ボランティア被験者のヒト尿中薬物スクリーニングを実施し、投与された医薬品化合物を同定して、それらを複雑な生物学的マトリックスの内因性化合物と区別しました。従来の UPLC-IM-MS ライブラリーと RGM-UPLC-IM-MS ライブラリーのアプリケーションを比較することで、このアプローチの実現可能性を評価しました。