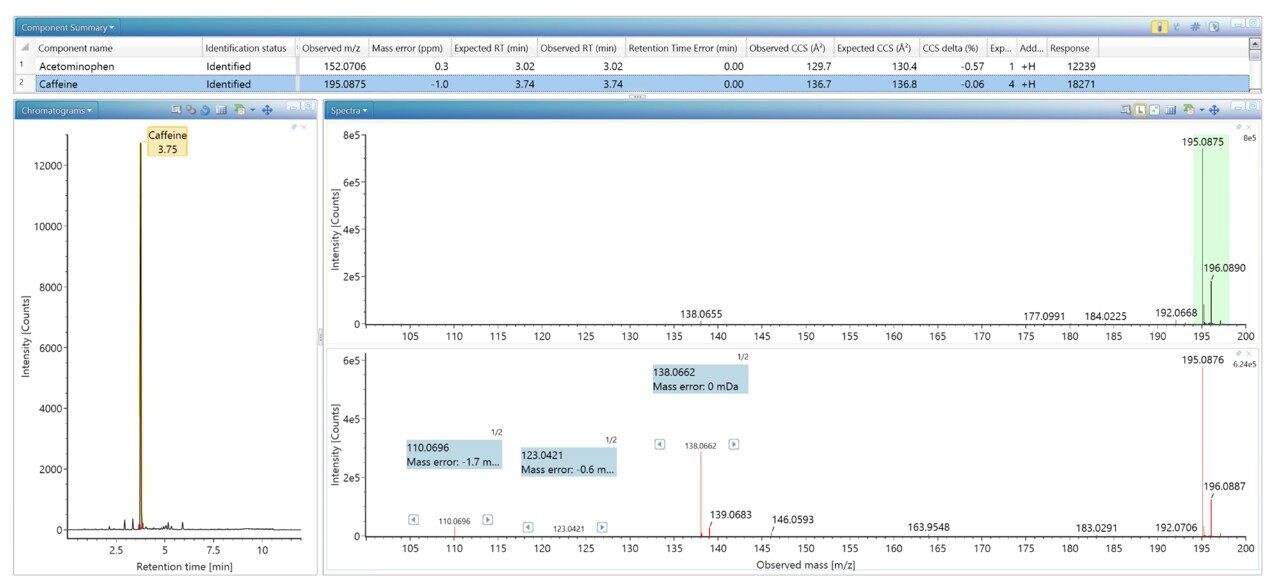
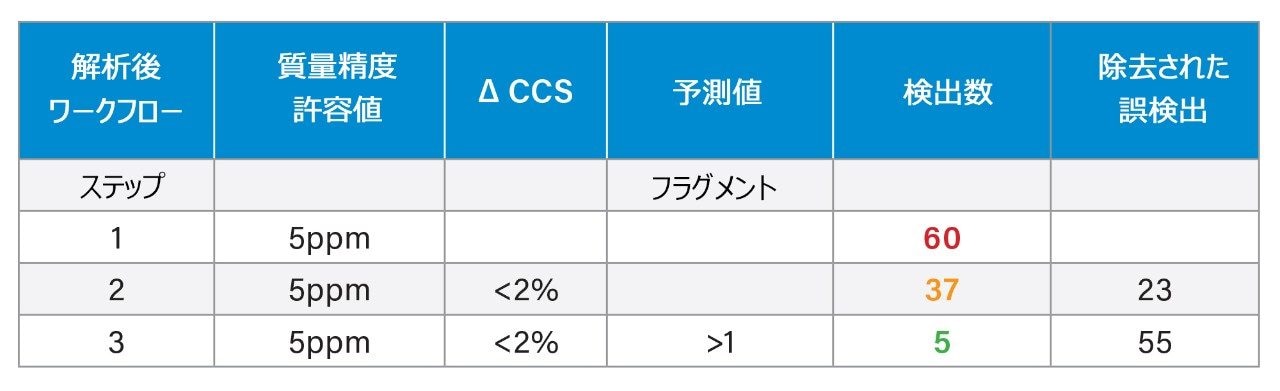
四重極飛行時間型質量分析計(Q-TOF)などの高分解能質量分析計(HRMS)は、尿や血液など、複雑な生体マトリックスに含まれる対象成分の臨床、法中毒学、代謝物構造推定におけるスクリーニングツールとして普及が進んでいます1,2。 ノンターゲット「フルスキャン」データ取り込みを使用することで、単一の分析で何千件もの検出を行い、続いて過去に遡ってターゲットデータ分析を行うことができます。世界的により高いサンプルスループットの向上が求められており、時間効率の向上やコスト削減への要求から、多成分化合物分析への移行が進んでいます。このアプローチは農薬、マイコトキシン、天然植物毒素3、有機汚染物質4,5 の分析に使用されていますが、これらは、食品6 から廃水などの環境サンプルに至るまで、複雑なサンプルマトリックス中にも含まれています7,8。スクリーニング法の目的は、調査対象サンプルに含まれるターゲット化合物を、誤検出率をできる限り最低限に抑えつつ、迅速に検出して同定することにあります。精密質量、同位体パターン、プロダクトイオンスペクトルなど、測定した化合物の特性を使用し、適切なフィルターを適用して、サンプル中における化合物の有無を判定しました。一方、複雑な生体マトリックス中に低濃度で存在する対象化合物については、これらの特性のみを使用してマトリックスや分析種を同定することは困難で、さらなる分析法開発戦略が必要になる可能性があります。このような複雑な分析では、新しい次元の IM 分離により、分析上の課題が軽減するとともに、衝突断面積(CCS)により追加の同定の特異性が得られます。
以前報告された質量分析ライブラリー作成戦略を使用し9、超高速高分離液体クロマトグラフィー-イオンモビリティー質量分析(UPLC-IM-MS)を用いて、市販 FDA 承認薬のセットを分析しました。使用する戦略により、保持時間(tr)、プリカーサーイオン、プロダクトイオン、CCS の決定が可能になります。IM 装置が製品化されたことにより(ウォーターズコーポレーション:SYNAPT(2006 年)、Vion(2015 年)、Cyclic IMS(2018 年))、査読済み論文の数が増加しました(2014/2015 年時点で 1,250 件以上)10-11。例えば農薬スクリーニングアッセイなどで、同定の特異性を補助する追加のエンドポイントとして CCS を使用する分析戦略が開発されています12。それ以来、低分子分析における CCS のルーチン使用が、医薬品(代謝、メタボロミクス、脂質)、食品安全性(動物用医薬品、マイコトキシン、ステロイド、天然物のスクリーニング、天然毒素)など、多様な分野にわたって増加しています。CCS で検索できるライブラリーが作成され、CCS の指標を使用して、同定における累積特異性を向上させるとともに誤検出を減らすことができます。天然物ライブラリーおよび食品添加物ライブラリーの作成については、CCS 測定の長期的頑健性および再現性の評価とともに、最近紹介しました14-17。
UPLC-IM は、イオンモビリティー(MS 分析前の気相分離)と UPLC(中性分子種の分離)の組み合わせで構成されています18,19。 UPLC(秒)、IMS(ミリ秒)、飛行時間型 MS(マイクロ秒)の時間スケールは、複雑なサンプルのハイスループット分析の要件に対応しています。化合物のイオンモビリティー分離は、質量分析部の前のガスを充塡した進行波イオンモビリティー(TWIM)RF イオンガイド内で、気相イオンが分離されることによって行われます。不活性バッファーガス(窒素)を介してイオンパケットを駆動させるか、比較的弱い電場を使用することで、移動度の分離を行います。イオンとバッファー気体の間の衝突の回数により、ドリフト時間の差が生じます。結果として得られる分離は、RF イオンガイドに沿った DC パルスの繰り返し印加に基づいています。イオンは周期的にパルスまたは波に追い越され、モビリティーが小さい分子種はモビリティーが大きい分子種より追い越される頻度が高くなります。このため、デバイスを通過する時間がモビリティーにより左右され、イオン質量、電荷、形状などの要因の関数になります。イオンモビリティーにより、LC(疎水性)および MS(m/z)に対して 3 次元目の分離が得られます。
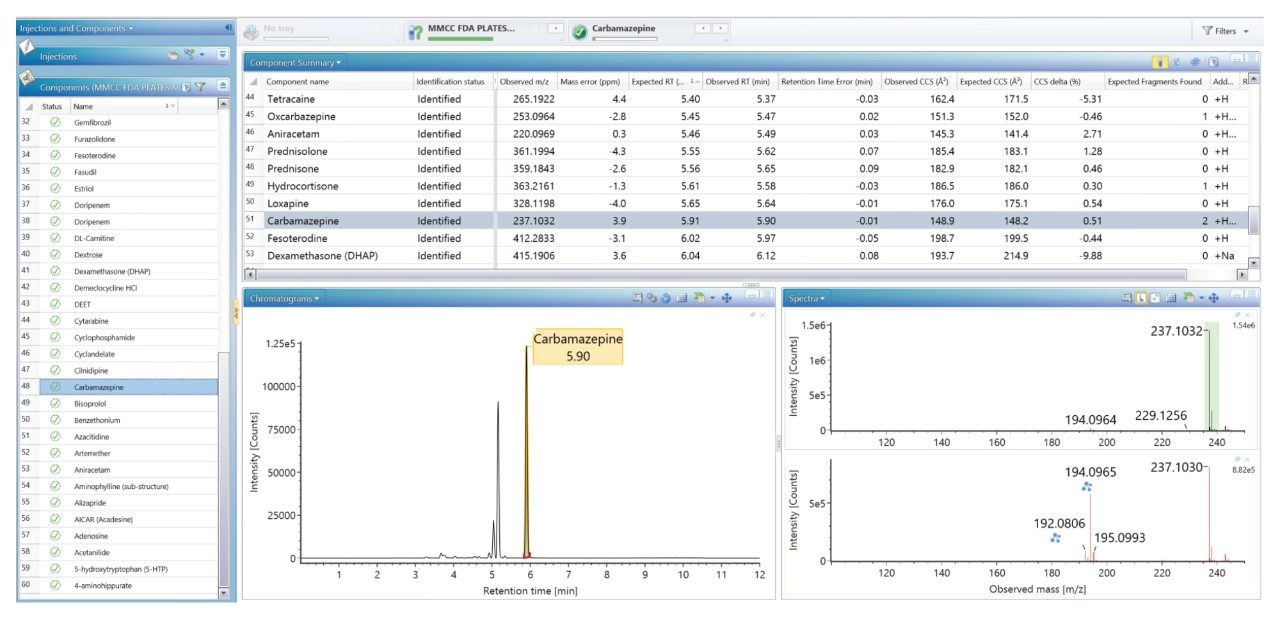
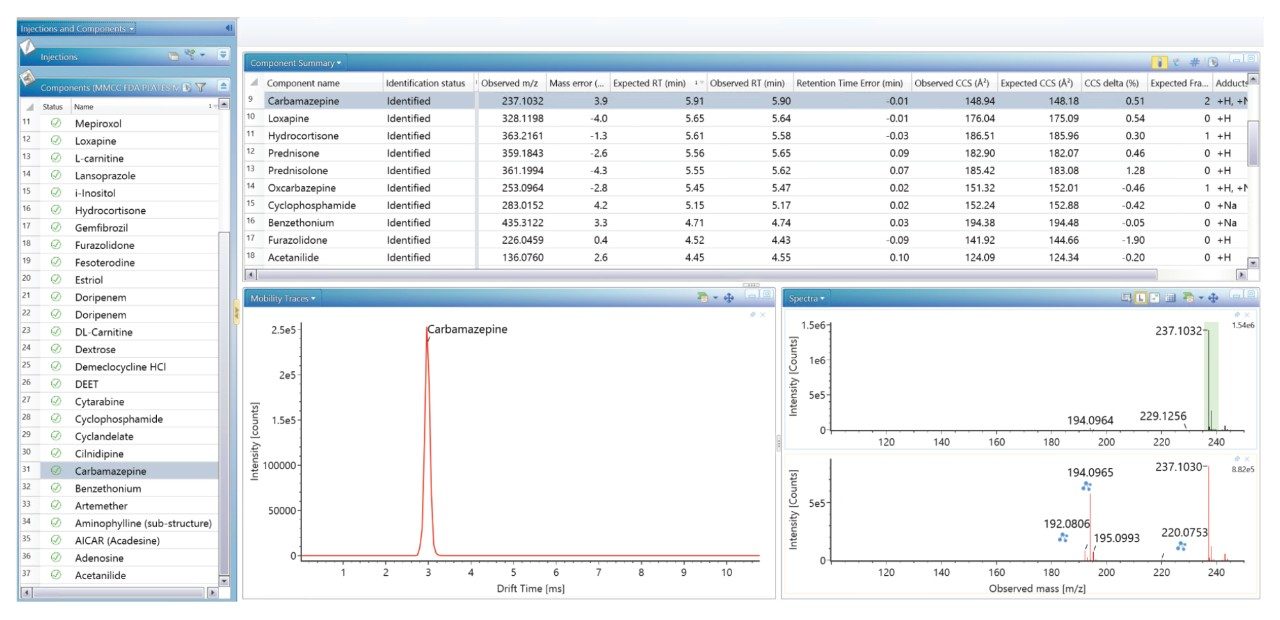
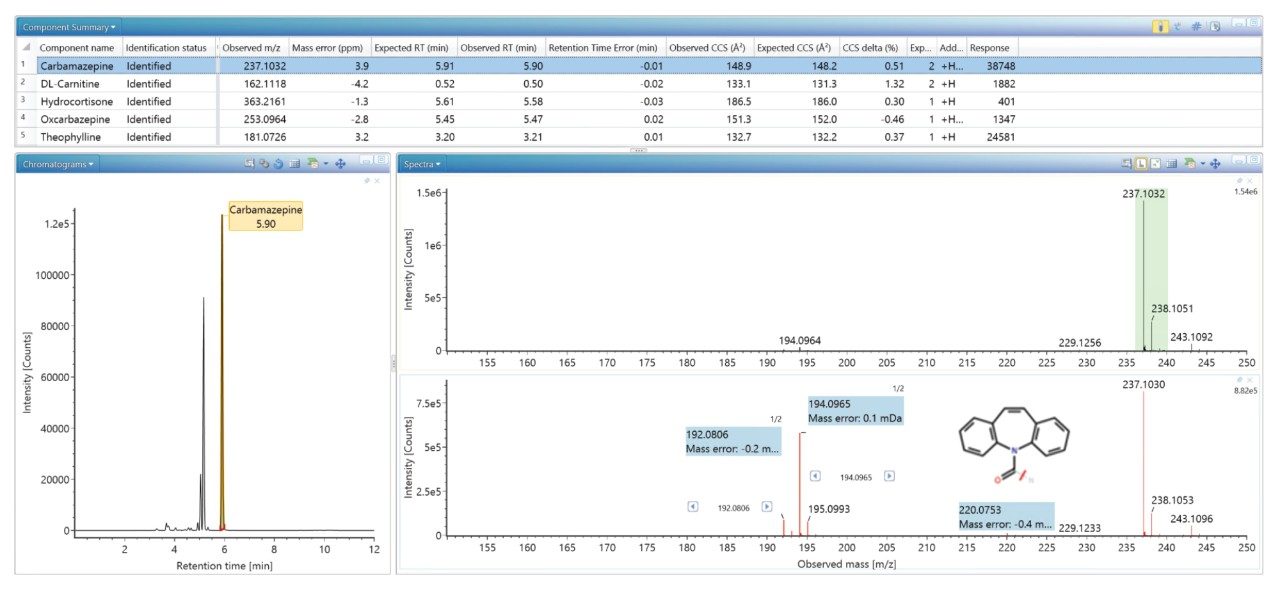
FDA が承認した低分子医薬品の大規模なライブラリーを生成し、これを使用して、患者サンプルのノンターゲット尿スクリーニングを実施し、投与した医薬品化合物を同定しました。