タンパク質のサイズ排除クロマトグラフィーを最適化するための迅速で体系的なメソッド開発アプローチ
要約
サイズ排除クロマトグラフィー(SEC)分析法は、望ましくない二次相互作用を緩和し、サイズに基づくエントロピー主導の分離が得られる条件を特定することを目標にして、頑健に開発する必要があります。タンパク質の場合、好ましくない静電相互作用および疎水性相互作用の両方が発生する可能性があります。新しい Premier Protein SEC カラムテクノロジーの基礎である表面により、この分離手法のより幅広い可用性が可能になります。それでもなお、新しい分析法を体系的に開発することは常に価値があります。本研究では、高低さまざまな状態の移動相バッファー強度、共溶媒濃度、カラム温度のスクリーニングを取り入れた 23 実験計画法(DoE)アプローチについて説明します。この 3 元配置実験計画法は、2 時間未満の装置実行時間で調査でき、その結果は単純なスプレッドシート形式の計算を使用して線形モデルに適合させることができます。2 種のモノクローナル抗体と 1 種の抗体薬物複合体の例が示されており、ユーザーの自らの分析法開発作業に役立つために、対応するスプレッドシートのテンプレートがオンラインで利用可能になっています。
アプリケーションのメリット
- SEC 分離を最適化するための迅速で体系的なアプローチ
- この方法を適用することにより、8 つの実験と 2 時間の実験作業のみで最適化した条件の決定が可能に
- 実験計画法のレスポンス関数として、凝集体回収率(HMWs%)、分離(Rs)、ピーク幅(w1/2)をモニター
はじめに
サイズ排除クロマトグラフィー(SEC)は、医薬品タンパク質の特性解析に過去から使用されてきた手法であり、(超遠心分析法(AUC)および動的光散乱(DLS)とともに)タンパク質凝集体の定性分析および定量分析の標準的な分析法と考えられています。この手法の利点の 1 つは、マイルドな非変性移動相条件を使用することです。ただし、この種のアプリケーションでは、SEC カラム内での非特異的相互作用に起因する困難な課題が存在する可能性があります。タンパク質分子種と、充塡剤およびカラムハードウェアとの間に望ましくない相互作用がある可能性があり、メソッドのパラメーターを特別に検討する必要があります1。 一部の汎用規則を分析法開発に適用でき、実験計画法(DoE)を使用することは、非特異的相互作用に対処するのに役立つ可能性があります。
SEC では、バルク移動相と充塡剤の内部ボリュームの間の分配効果は、物理化学的吸着がなく、エントロピー過程によって理想的に機能します。ただし実際には、これが達成されることはめったにありません。生体高分子(モノクローナル抗体(mAb)ベースの医薬品など)は、さまざまな程度の疎水性および静電特性の多面的な表面を呈します。このため、充塡剤とクロマトグラフィーハードウェア(カラムフリットなど)の両方と疎水性および静電性の二次的相互作用の可能性があり、ピーク回収率、ピーク形状、分析法の頑健性に悪影響を及ぼすことがあります。このような望ましくない二次的相互作用を緩和するため、分析法のパラメーターを慎重に最適化する必要があります。
(1)不活性カラムハードウェアの使用、および(2)分析法パラメーターの最適化は、2 つの重要な検討事項です。後者については、移動相中の塩および有機モディファイヤーの濃度、カラム温度、流速を調整することで、分析法の機能を微調整できます2,3。 SEC 分析法の開発については、最初のステップは、適切な充塡剤の選択になります。一般的に、対象分析種の 2 ~ 3 倍の平均孔径の充塡剤を選択します。例えば、ほとんどの mAb は平均水和半径がほぼ 50 Åです。さらに、mAb 凝集体の水和半径は 100 Å 程度になることがあります。そのため、mAb の SEC 分離に最も適した平均孔径は一般に 200 Å ~ 300 Å です。さまざまな分子種の溶出量とそれらの間の選択性は、充塡剤の孔径と孔径の分布によってのみ、最適に決まります。一方、非特異的相互作用が発生すると、溶質が保持され、幅の広い非対称のピークとして溶出して、分離と凝集体の定量の両方が損なわれる可能性があります。
静電相互作用を低減するには、移動相のイオン強度または塩濃度を増加するのが一般的です4,5。 有機モディファイヤー(メタノール、エタノール、アセトニトリルなど)を使用しても、疎水性相互作用の強度を下げることができ、多くの場合 5 ~ 10% の有機ディファイヤーを添加することでピーク形状が改善されます6。 SEC での平衡はエントロピーに依存しているため、温度は平衡定数(K)にほとんどまたはまったく影響せず、このためにゾーン保持係数(k”)にも影響しません。(従来の保持係数(k または k’)は SEC には適用できないことに、注意してください。代わりに k” が使用されます。これはポア内に留まっている移動相、または移動しているバルク移動相の中の、サンプルの確率の比を表します。つまり、k” は内部ポアと間隙容量の間の溶質の分布を意味します)。平衡定数は式 1 に従って定義できます。
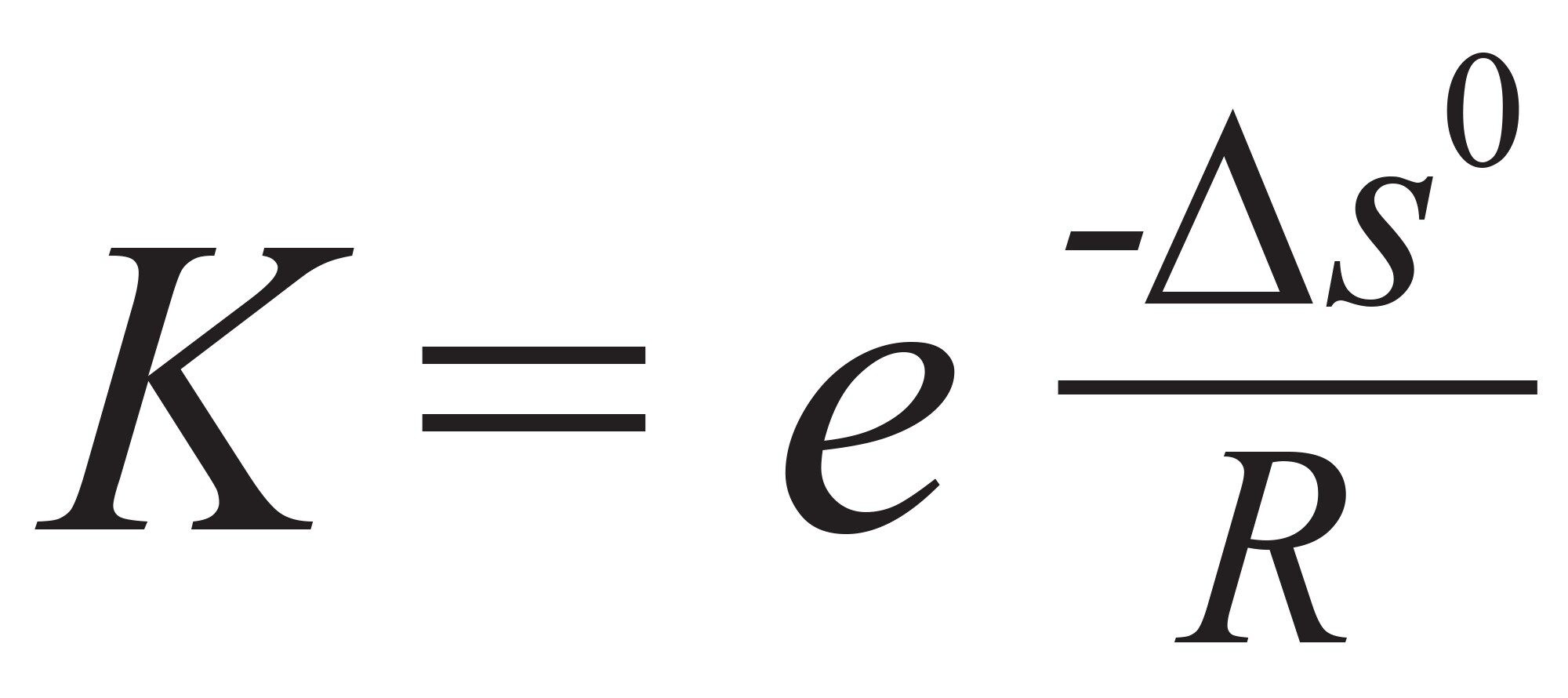 式 1
式 1
ここで ΔS0 は化学的転移時のエントロピー変化、R は一般ガス定数です。二次的相互作用が発生した場合、その強度は温度によって異なる可能性があるため、温度は回収率やピークテーリングに影響を及ぼす(改善する)有用な変数(要因)になる可能性があります。実際に、温度は、分析法の最適化、特に分析法のコントロールと再現性に関して考慮する重要な要素である場合があります。
SEC 分析法開発は、多くの場合時間がかかり、通常は試行錯誤のアプローチに基づきます。本アプリケーションノートでは、3 つの要因(塩濃度、有機モディファイヤー濃度、移動相温度)の効果とそれらが SEC 分離の品質に及ぼす影響を試験するためのシンプルな DoE が提案されています。最適な動作条件を迅速に定義するための体系的なアプローチが提供されます。
提案された要因(xj)およびそのレベル(+/-)
1)移動相の塩濃度、例えば、リン酸緩衝生理食塩水(PBS):1x PBS(x1(+1))、2 倍希釈(1/2x)PBS バッファー(x1(-1))(Cold Spring Harbor プロトコルに従って調製)7。 PBS は現在、水系 SEC 分離用のバッファーに、ルーチンに使用されています。ただし、一般に使用されるリン酸カリウム、リン酸ナトリウム、酢酸アンモニウム組成のような、その他のバッファーも使用できます。この場合、DoE の推奨バッファー濃度レベルは 50 mM(x1(-1))および 150 mM(x1(+1))です。
この要因(x1)を活用して、静電相互作用を詳しく調査できます。静電性の二次的相互作用は、低塩濃度移動相では HMWs% の減少として現れる傾向があります。高塩濃度でのモノマー分子種とダイマー分子種の間の HMWs% の向上および選択性の増加は、静電相互作用の緩和を示しています。
2)移動相での有機モディファイヤー(MeOH、ACN、EtOH、iPrOH など)の濃度(例えば、2%(x2(-1))および 10%(x2(+1))。
この要因(x2)を利用して、疎水性相互作用を詳しく調査できます。疎水性相互作用は、HMWs% の減少、ピークテーリング、または溶出時間のシフトとして観察される傾向があります。有機モディファイヤーの添加により HMWs% の増加、よりシャープなピーク、溶出時間の早まりが発生する傾向があり、完全水系条件下で疎水性の二次的相互作用が起こっている可能性があります。
3)移動相温度(例えば、25 ℃(x3(-1))および 35 ℃(x3(+1))は、変性を避けるために適切な範囲に保つ必要があります。
この要因(x3)を活用し、タンパク質の立体構造と拡散性、および充塡剤表面の溶媒和を変えることができます。さらに、非特異的相互作用の強度を下げて、分離品質を向上できます。
これらを分類的(定性的)要因(xq,j)と考えて、さまざまなカラムも比較できます。
充塡剤ケミストリー、ハードウェアの種類、平均孔径、孔径の分布のすべてが、SEC 分離の品質と HMW 分子種の回収率に影響を及ぼします。これらそれぞれは分類的(不連続)要因と見なすことができます。
モニターするレスポンス(yj)の提案
保持クロマトグラフィーモードでは、これは溶質保持であり、多くの場合最適化されます。市販のさまざまなソフトウェアパッケージは、十分に確立された半経験的保持モデルに基づく保持のモデリングに役立ちます。ただし、SEC では溶質は保持されず、保持および選択性のモデリングは行えません。保持のモデリングの代わりに、DoE を適用して最も重要な要素の効果を詳しく調べる方が、より現実的と思われます。
SEC の場合、全般的に最も重要なレスポンスは次記の 3 つです:
1)モノマーと HMWsの間のRs(y1)、
2)HMWs%(y2)、
3)メインピーク(モノマー)の幅(y3)
ピークの対称性とベースラインに戻るまでの時間も、多くの場合レスポンスと見なされます。ただし、ほとんどの場合、これらは HMWs% 回収率およびピーク幅と非常によく相関しています。これは、これらのレスポンスのそれぞれが非特異的な二次的相互作用の尺度であるためです。HMWs% 値が高い場合やピークがシャープな場合は、ピーク対称性も許容できるはずです。このため、最初のステップとして、上記のレスポンスのみを検討することを推奨します。
DoE の提案
23 完全要因計画(線形モデル)が提案されていますが、要因計画やプラケットバーマン計画、または非線形モデル用ボックスベンケン計画など、他の計画も適用できます。図 1 に、提案された 23 計画および対応する計画マトリックスの概略ビューが示されています。
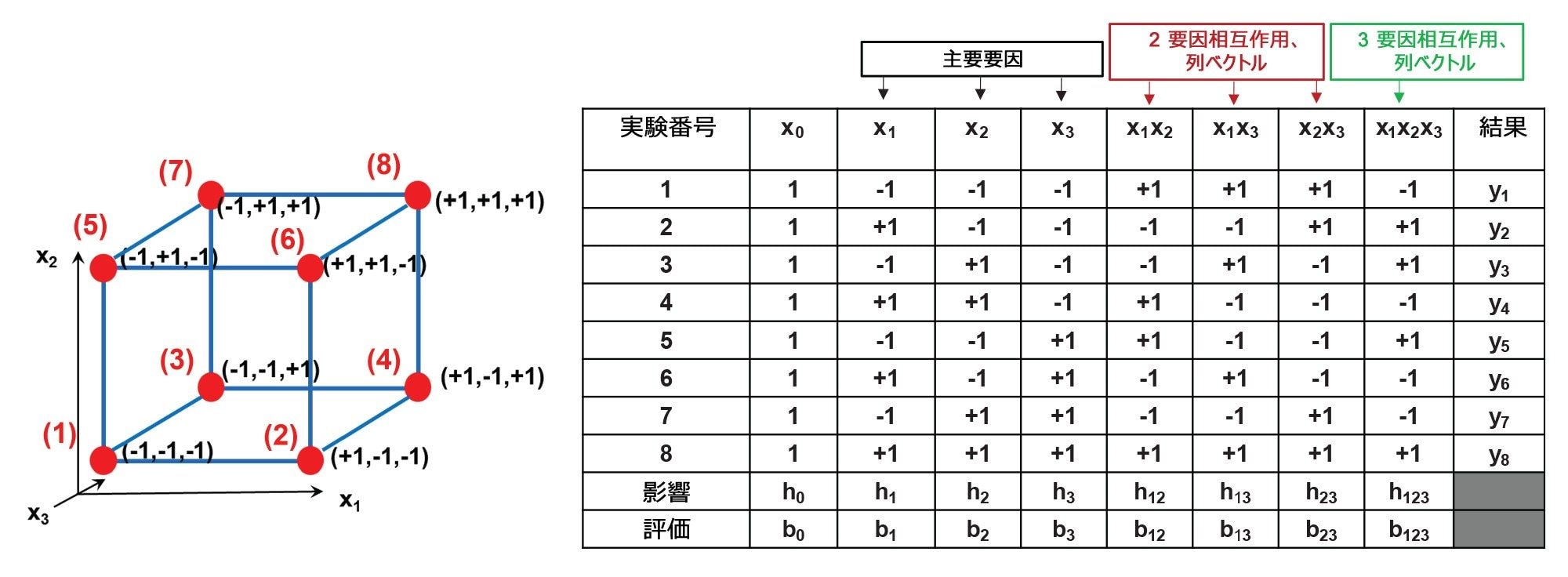 図 1. 23 完全要因計画(左)および関連する計画マトリックス(右)の概略図。x1 ~ x3 は選択された要因であり、立方体の頂点にある赤色の点(1 ~ 8)は条件、および 8 つの実験の対応する要因のレベル(+1 または -1)を示します。y1 ~ y8 は実験で測定されたレスポンス、b0 ~ b123 は算出されたモデルパラメーターです。
図 1. 23 完全要因計画(左)および関連する計画マトリックス(右)の概略図。x1 ~ x3 は選択された要因であり、立方体の頂点にある赤色の点(1 ~ 8)は条件、および 8 つの実験の対応する要因のレベル(+1 または -1)を示します。y1 ~ y8 は実験で測定されたレスポンス、b0 ~ b123 は算出されたモデルパラメーターです。
いくつかの市販のソフトウェアパッケージにより、DoE のモデルパラメーターおよび要因の効果を直接計算できます。ここでは、モデルおよび要因の影響を手動で計算することを推奨します。この作業は、Microsoft® Excel® またはその他のスプレッドシートソフトウェアで簡単に行えます。
要因 j の影響(h)は次記の式で計算できます:式(2)
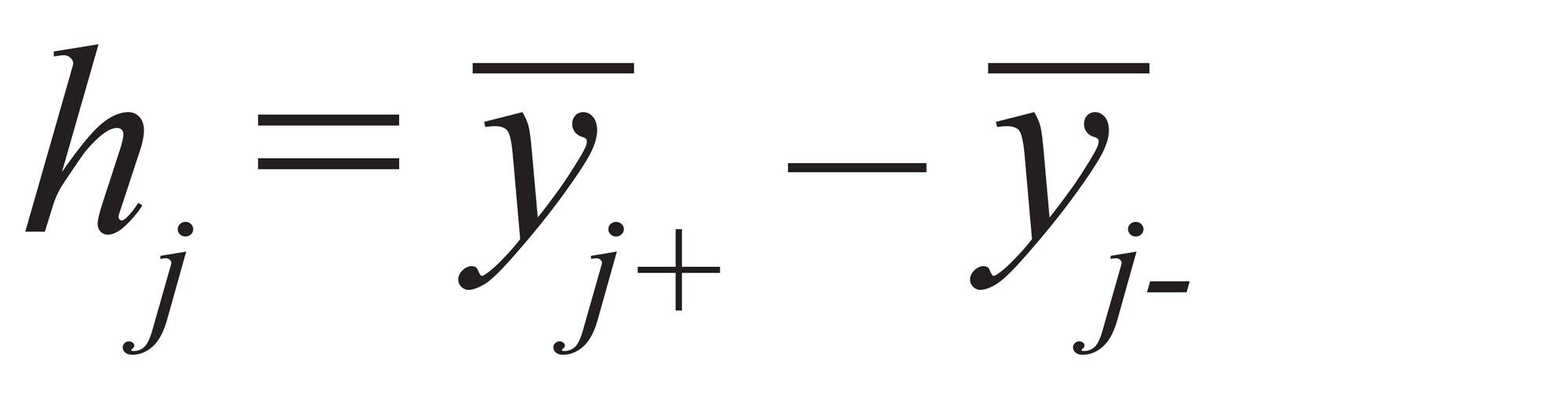 式 2
式 2
ここで、yi+ は要因 j の +1 レベルで測定されたレスポンスの平均で、yi- は要因 j の -1 レベルで測定されたレスポンスの平均です。
要因 j のモデルパラメーター(b)は式(3)のとおりです。
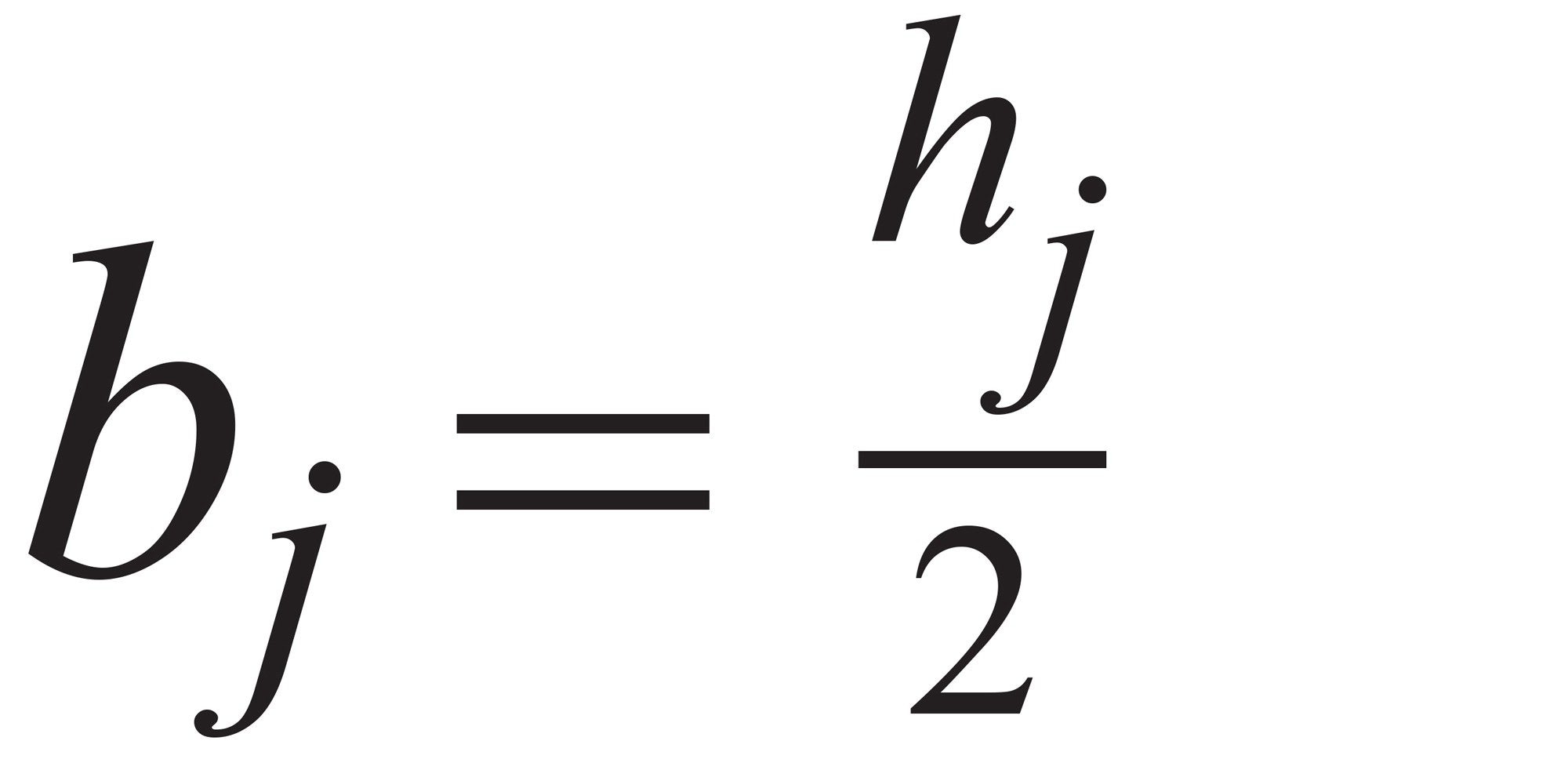 式 3
式 3
b0 パラメーターは、測定されたすべてのレスポンスの平均です:式(4)
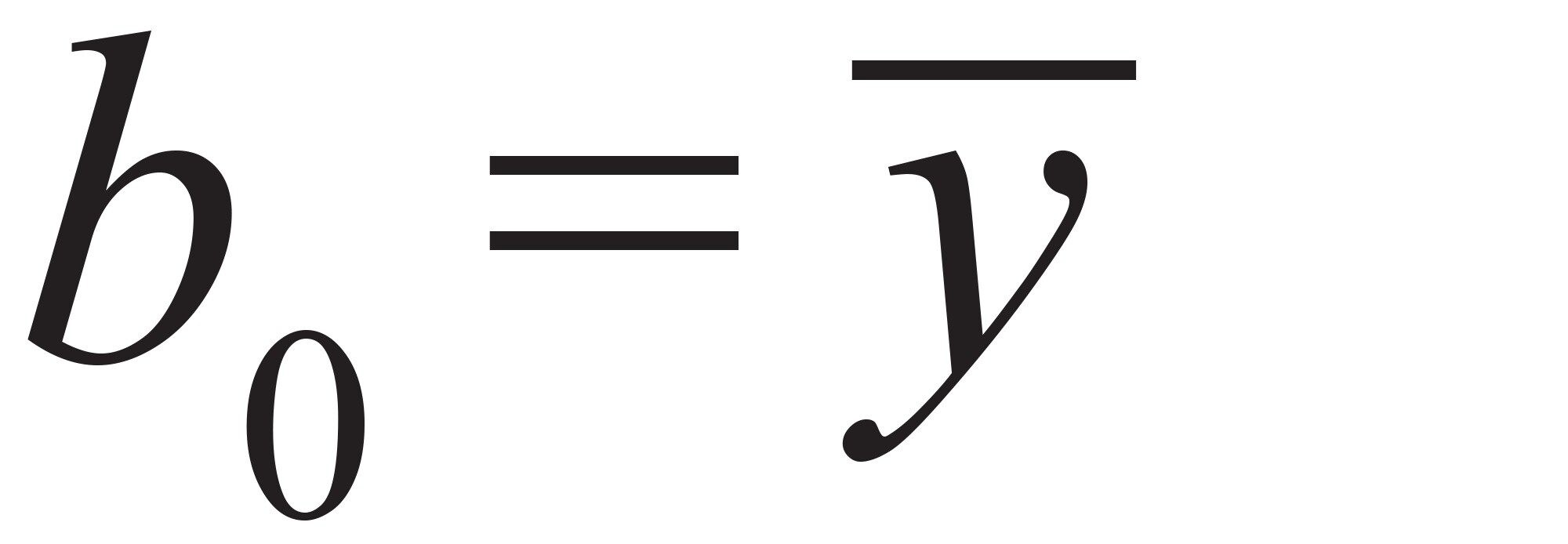 式 4
式 4
次に、以下のモデルを書き込んで、任意の条件でのシステムのレスポンスを推定できます。
主要な相互作用のみが考慮されます(1 次線形モデル):式(5)
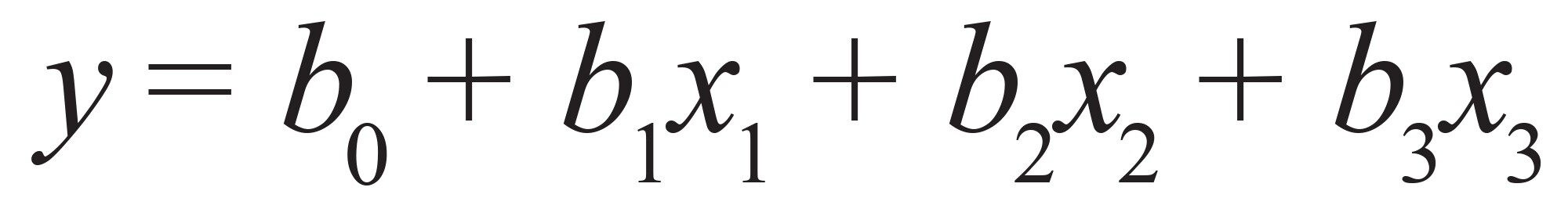 式 5
式 5
要因の相互作用を考慮する場合(3 次線形モデル):式(6)
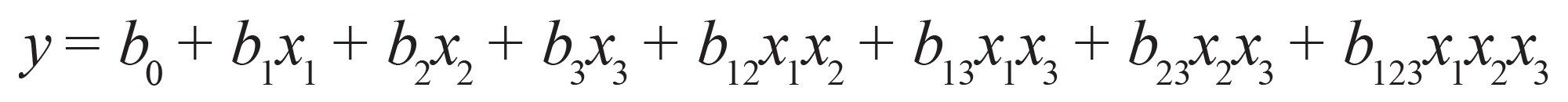 式 6
式 6
最終的に、要因の値は要因レベルに変換できます。zj が任意の xj 要因の対応する設定値である場合、式(7)
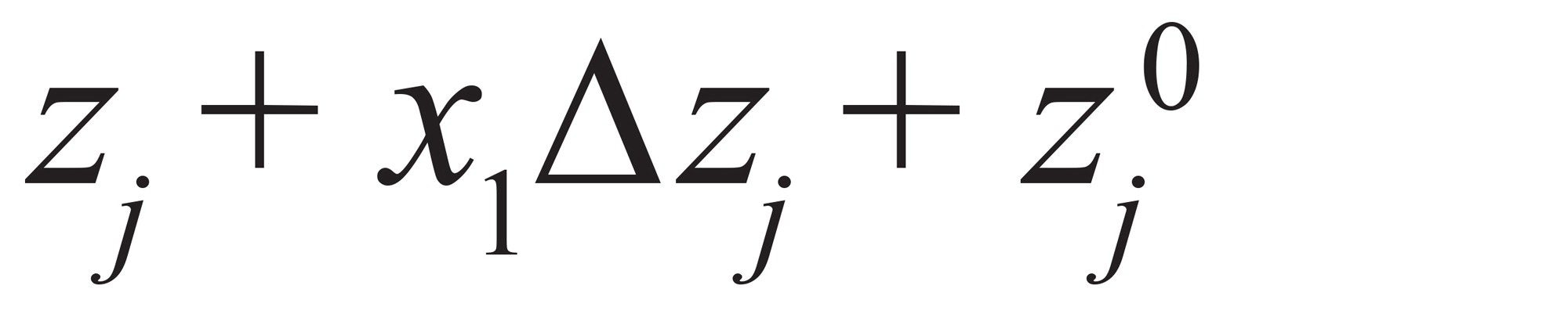 式 7
式 7
ここで、Δzj は要因 j の 1 レベルの差で、z0jは要因 j の平均値です。
したがって、要因 j のレベルは次の式で計算できます:式(8)
 式 8
式 8
最後に、要因の影響をパレート図で表示でき、式 5 または式 6 を使用して、要因レベルの任意の組み合わせについてシステムレスポンスを予測できます。空白の Excel スプレッドシートをすばやく準備できます。
実験方法
サンプルおよび移動相の調製:
リツキシマブ(リツキサン)は Biogen MA Inc.(米国マサチューセッツ州ケンブリッジ)から、カドサイラ(トラスツズマブエムタンシン)とアバスチン(ベバシズマブ)は Genentech Inc.(米国カリフォルニア州サンフランシスコ)から購入しました。サンプルを水で 1 mg/mL に希釈し、それ以上の調製を行わず、直接注入しました。HPLC グレードの水およびメタノール(MeOH)は、Fisher Scientific(アイルランド、ダブリン)から入手しました。リン酸緩衝生理食塩水(PBS)の成分(塩化カリウム(KCl)、塩化ナトリウム(NaCl)、リン酸水素二ナトリウム(Na2HPO4)、リン酸二水素カリウム(KH2PO4))は Sigma-Aldrich (スイス、Buchs)から購入しました。
LC 条件
|
LC システム: |
ACQUITY™ H-Class Bio Plus(クオータナリー) |
|
検出: |
280 nm での UV 検出 |
|
バイアル: |
ポリプロピレンバイアル(製品番号:186002639) |
|
カラム: |
XBridge™ Premier Protein SEC カラム、250 Å、2.5 µm、4.6 × 150 mm(製品番号:186009959) |
|
カラム温度:(要因 3、x3): |
25 ℃(-1 レベル)および 35 ℃(+1 レベル) |
|
サンプル温度: |
8 ℃ |
|
注入量: |
1.0 µL(サンプル) |
|
流速: |
0.3 mL/分(分析時間は 8 分に設定) |
|
移動相: |
pH 7.4 1x PBS および 1/2x PBS)、コールドスプリングハーバーのプロトコル [7] に従って調製(2% または10% MeOH を含む)。 |
|
(要因 1(x1)および要因 2(x2)): |
x1 要因のレベル:1/2x 強度の PBS(希釈係数 2、-1 レベル)、1x 標準レシピの PBS(希釈係数 1、+1 レベル) x2 要因のレベル:2% MeOH(-1 レベル)、10% MeOH(+1 レベル) |
実験条件は、図 2 に 23 要因 DoE として示されています。
 図 2. 移動相の塩濃度(x1 要因、PBS バッファーの希釈係数)、移動相の MeOH%(x2 要因)および移動相の温度(x3 要因)の影響を推定するために DoE に設定した、8 つの実験条件。
図 2. 移動相の塩濃度(x1 要因、PBS バッファーの希釈係数)、移動相の MeOH%(x2 要因)および移動相の温度(x3 要因)の影響を推定するために DoE に設定した、8 つの実験条件。
結果および考察
8 つの実験を行い、以下のレスポンスが登録されました(リツキシマブに対して):
- モノマーと HMWs の間の Rs(y1)、
- HMWs%(y2)、
- メインピーク(モノマー)の幅(y3)
3 つのレスポンスすべてについて計画マトリックスに記入し、要因の影響を推定しました。図 3 に、代表的な例として y2 レスポンス(HMWs%)のマトリックスが示されています。
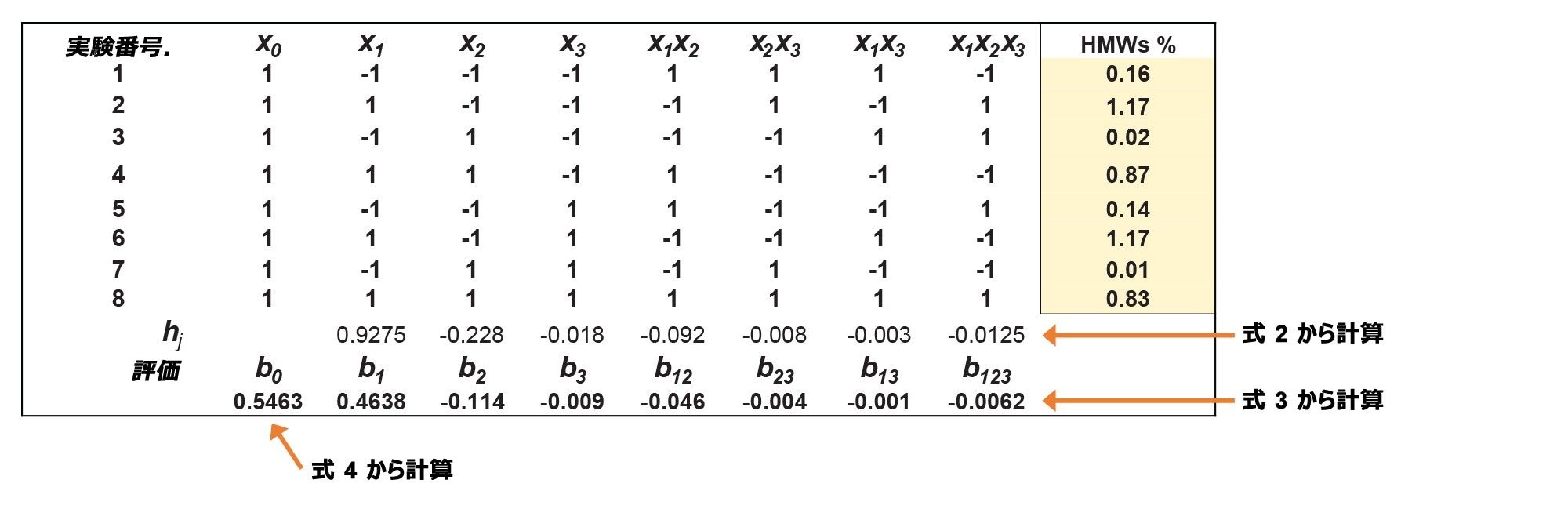 図 3. y2 レスポンスの DoE マトリックス(測定した HMWs%)
図 3. y2 レスポンスの DoE マトリックス(測定した HMWs%)
要因の影響およびモデルパラメーターをマトリックスから決定しました。例として、y2 要因について、式 5 および式 6 に基づいて以下のモデルが得られました:
主要な相互作用のみを考慮した場合(式5):式(9)
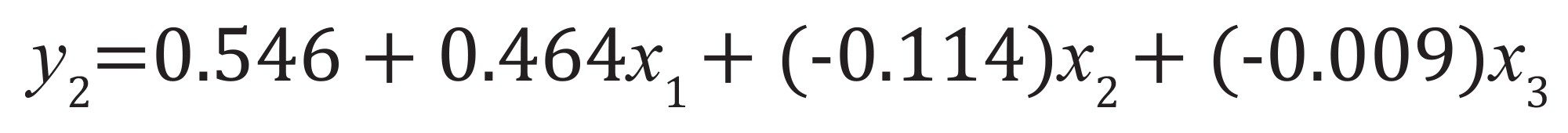 式 9
式 9
要因の相互作用を考慮した場合(式6):式(10)
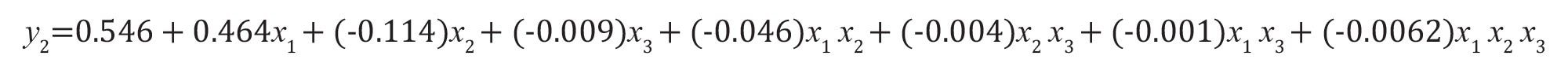 式 10
式 10
これらのモデル式を使用することで、(許容範囲内で)分析法要因の任意の組み合わせに対してレスポンスを推定できます。例えば、DoE の中心点の y2 要因(HMWs%)の予想値(x1 = 0 [z1 = 1.5x PBS]、x2 = 0 [z2 = 6% MeOH]、x3 = 0 [z3 = 30 ℃])は、y2pred = 0.546% HMWs と予測されます。実験的な検証を行い、y2exp = 0.550% HMWs が得られましたが、これは HMWs% 予想値の -0.7% 誤差に相当します。(要因レベルと要因値の間の変換は、式7 および 8 で行うことができます。)HMWs% の予測値の偏差がこのように小さいことにより、線形モデルが妥当であることが示されています。
要因の影響をパレート図としてプロットすることは、非常に有用です。図 4 に、y2 レスポンスについて得られたパレート図が示されています。見てわかるように、観察された HMWs% には x1 要因(移動相の PBS 濃度)が最も大きな影響を及ぼし、HMWs 分子種と固定相(またはカラムハードウェア)の間にいくらかの静電性二次的相互作用が存在することが示唆されています。移動相の MeOH% は HMWs% に小さな影響を与え、温度と要因の相互作用の影響は無視できます。
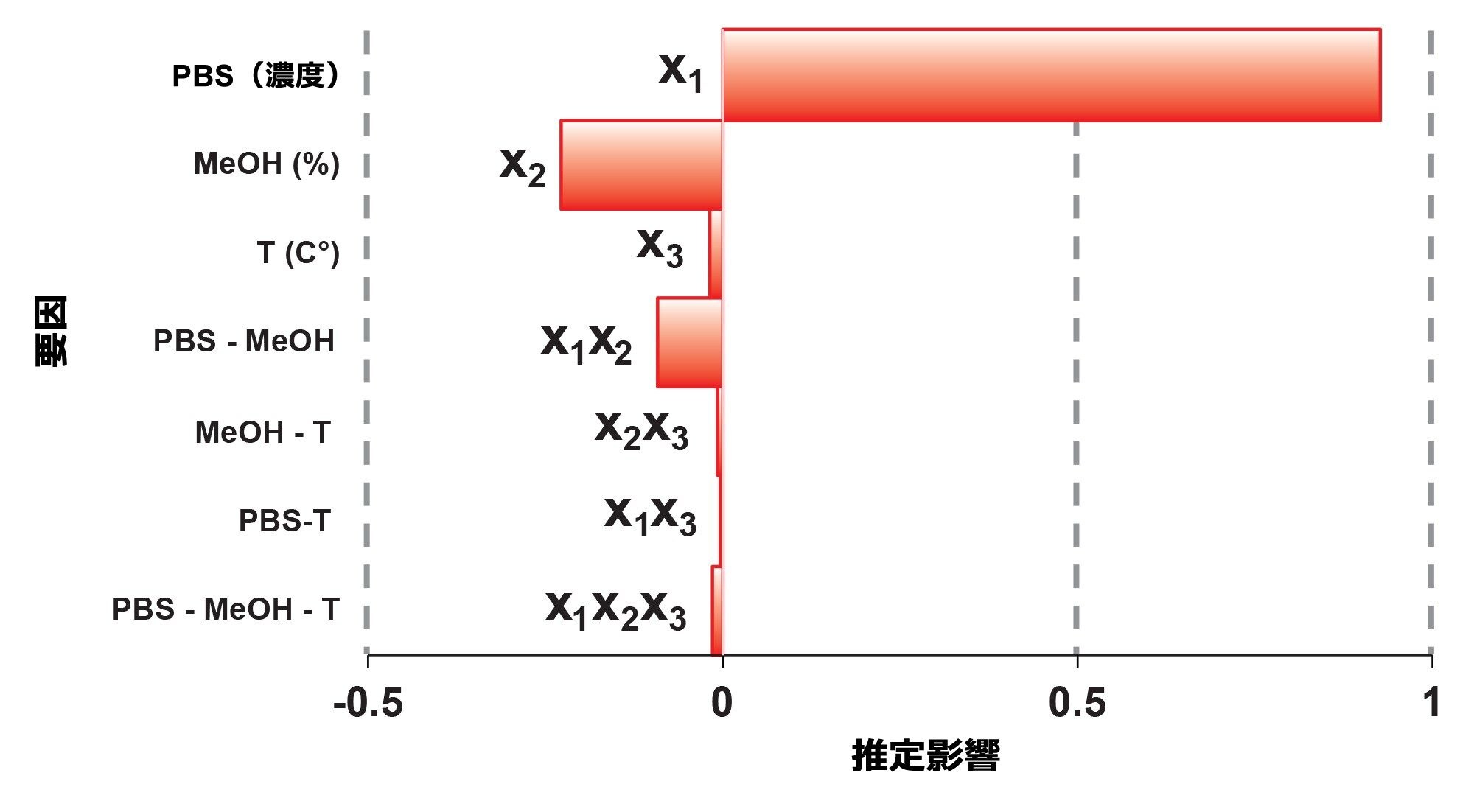
y1(Rs)および y3(ピーク幅)のレスポンスにも同じアプローチを使用しました。いずれのレスポンスも x1 要因(移動相の PBS 濃度)の影響を受け、y3 も移動相温度の影響を大きく受けました。最終的に、この SEC 分離に 1x PBS バッファー、0% MeOH、35 ℃ を設定して、最も高い回収率と最もシャープなピークを得ました。
図 5 A に、選択したワーキングポイント(最適)に対応する条件で得られたクロマトグラム、図 5 B に最悪の条件(HMWs% 回収率が最も低く、メインピークの対称性が最も悪い)で得られたクロマトグラムが示されています。
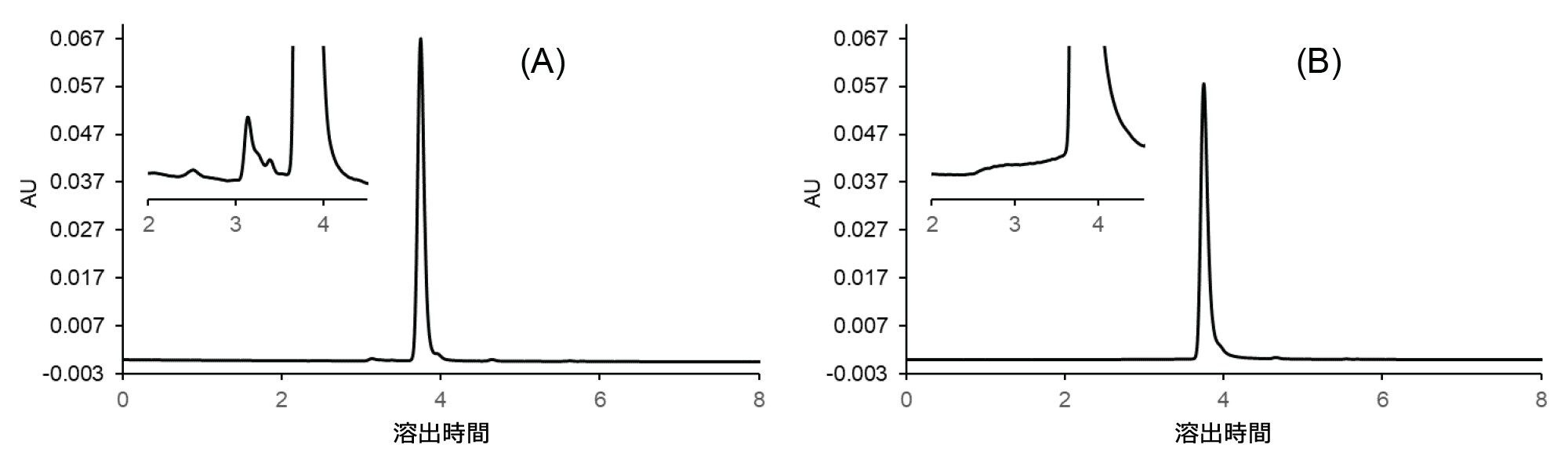 図 5. リツキシマブサンプルの実験的に測定した SEC クロマトグラム:XBridge Premier Protein SEC 250 Å 2.5 µm 4.6 × 150 mm カラムで得られた最適条件(A)および最悪条件(B)
図 5. リツキシマブサンプルの実験的に測定した SEC クロマトグラム:XBridge Premier Protein SEC 250 Å 2.5 µm 4.6 × 150 mm カラムで得られた最適条件(A)および最悪条件(B)
最適のワーキングポイントを見つけたら、分析時間を短縮するために流速を調整できます。内径 4.6 mm の SEC カラムでは、流速 0.3 mL/分で分析時間と効率のバランスが良好であり、初期の DoE 実験でこの流速を使用した理由がこれによって説明されています。それでもなお、対象ピークの間の分離と感度が十分に高い場合は、より速い流速を検討することができます。流速は、SEC 分離の動的性能とピークの光学検出の感度にのみ影響します。SEC の性質により、高流速では低効率になることが予想されます。逆に、初期の DoE 実験で分離が許容値に達していない場合は、流速を減らして分離を改善できます。図 6 に、流速 0.7 mL/分を使用したリツキシマブの SEC 分離の加速が示されています。HMWs 分子種とメインピーク(モノマー)の間の分離はこれでも許容可能なままであり、HMWs% の測定量は、流速 0.3 mL/分の場合の結果と変わっていませんでした。
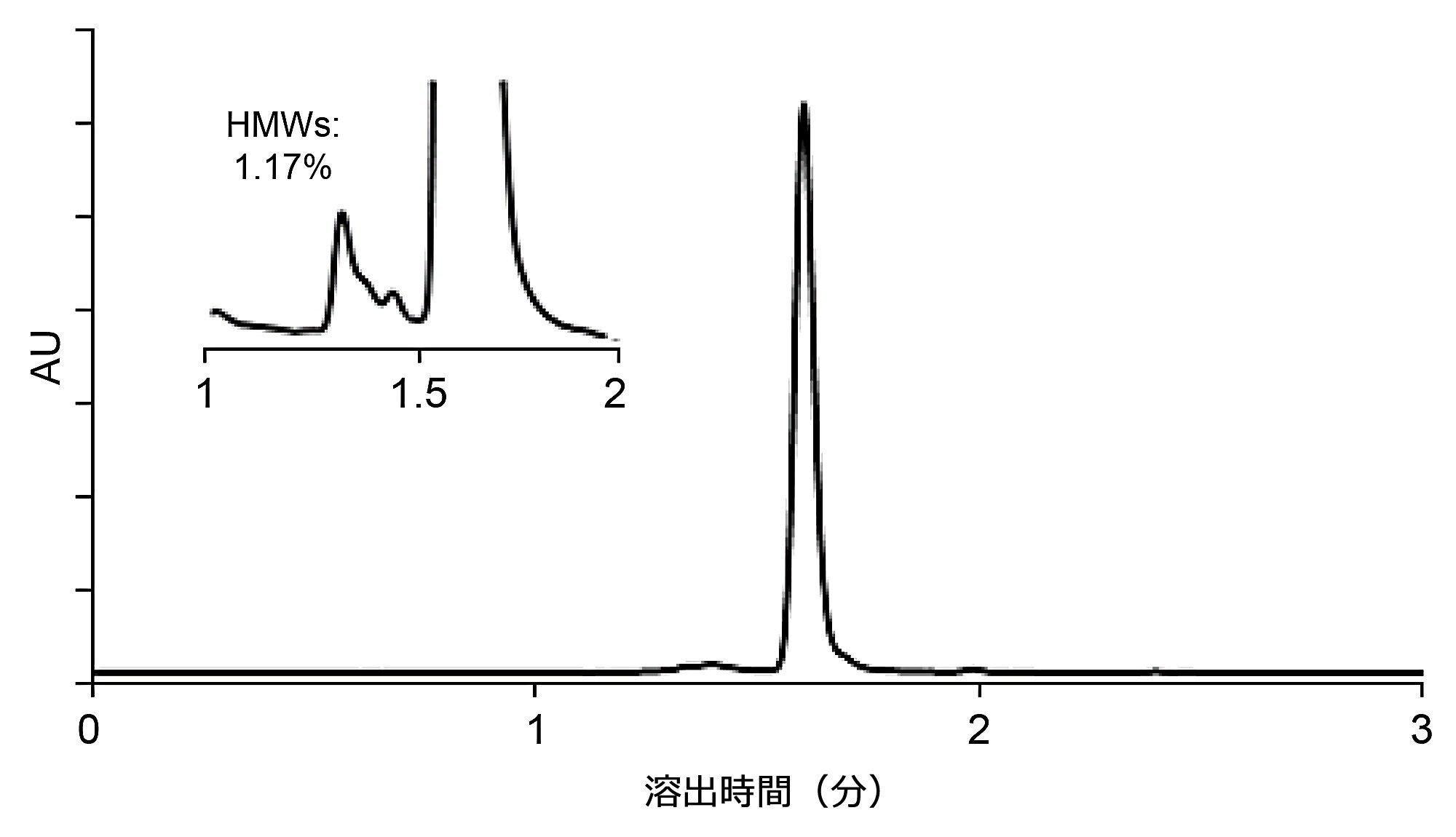 図 6. XBridge Premier Protein SEC 250 Å 2.5 µm 4.6 × 150 mm カラムで行った高速 SEC 分離(リツキシマブ)の例。移動相:1x PBS (pH 7.4)、流速 F = 0.7 mL/分、温度 T = 35 ℃、注入量 1 µL、検出:280 nm (UV)。
図 6. XBridge Premier Protein SEC 250 Å 2.5 µm 4.6 × 150 mm カラムで行った高速 SEC 分離(リツキシマブ)の例。移動相:1x PBS (pH 7.4)、流速 F = 0.7 mL/分、温度 T = 35 ℃、注入量 1 µL、検出:280 nm (UV)。
同様の DoE 最適化を、抗体薬物複合体トラスツズマブエンタンシンおよび mAb ベバシズマブに適用しました。ワーキングポイントを見つけた後、流速を 0.7 mL/分に増加しました。最終的なクロマトグラムが図 7 に示されています。
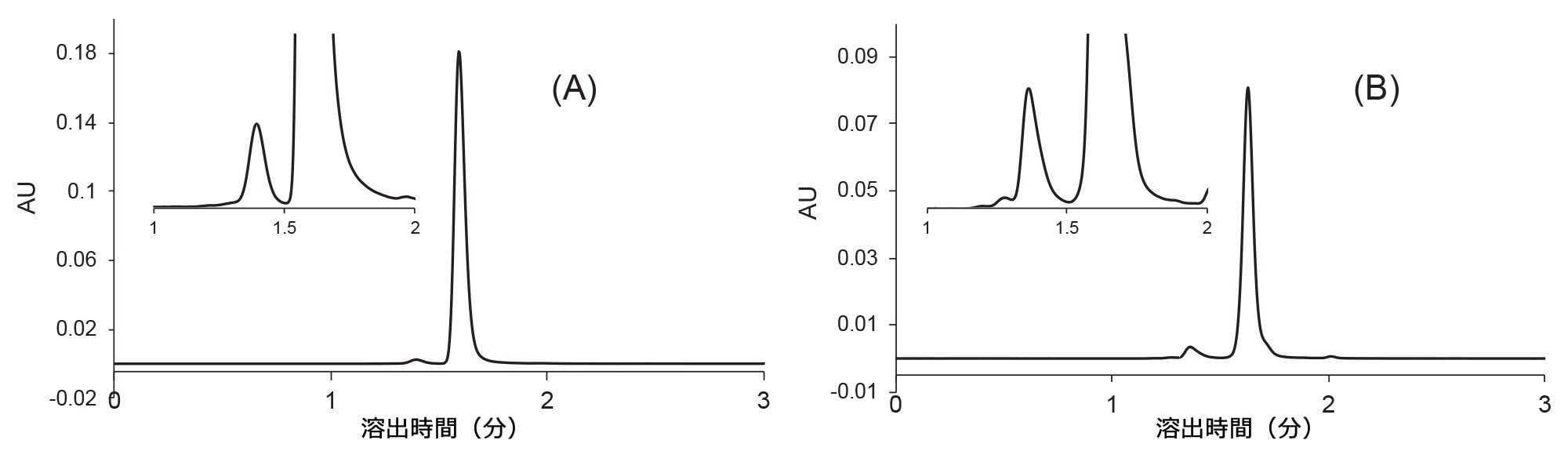 図 7. XBridge Premier Protein SEC 250 Å 2.5 µm 4.6 × 150 mm カラムを用いた、トラスツズマブエンタンシン(A)およびベバシズマブ(B)の高速 SEC 分析。移動相:1x PBS (pH 7.4)、流速 F = 0.7 mL/分、温度 T = 25 ℃、注入量 1 µL、検出:280 nm(UV)。
図 7. XBridge Premier Protein SEC 250 Å 2.5 µm 4.6 × 150 mm カラムを用いた、トラスツズマブエンタンシン(A)およびベバシズマブ(B)の高速 SEC 分析。移動相:1x PBS (pH 7.4)、流速 F = 0.7 mL/分、温度 T = 25 ℃、注入量 1 µL、検出:280 nm(UV)。
結論
医薬品タンパク質の SEC 分離を最適化するための体系的で迅速な分析法開発アプローチが提案されています。SEC では、分析種の保持がないはずであり、一般的な保持モデリングアプローチは適用されません。多くの場合に使用される半経験的保持モデルではなく、今回、移動相塩濃度(x1)、移動相有機モディファイヤー濃度(x2)、移動相/カラム温度(x3)など、分離に最も関連する要因の影響を試験するための、23 完全要因 DoE アプローチを提案しました。レスポンスを慎重に選択し、モノマーと HMWs の間の Rs(y1)、測定された HMWs%(y2)、メインピークの幅(y3)が最も意味のあるレスポンスであることを提案しました。上記の要因とレスポンスを試験することにより、静電性二次的相互作用および疎水性二次的相互作用の両方について詳しく調査し、理解して、最終的に緩和することができます。
最終的に、簡単な線形モデルによって、要因レベルの任意の組み合わせについてレスポンスを予測でき、最適なワーキングポイントを特定できます。
4.6 × 150 mm 構成の低吸着カラムテクノロジーを適用し、2.5 µm 粒子を充塡することで、この迅速な分析法開発手順を数時間内に完了できます。さらに、このアプリケーションノートに記載されている式と一般に入手可能なスプレッドシートテンプレートを使用して、簡単に独自の DoE ベースの分析法開発戦略を再作成および設計できます。スプレッドシートのテンプレートはこちらからダウンロードできます。
参考文献
- P. Hong, S. Koza, E.S.P. Bouvier, A Review, Size Exclusion Chromatography for the Analysis of Protein Biotherapeutics and Their Aggregates, J. Liq.Chromatogr. Relat.Technol.35 (2012) 2923–2950.https://doi.org/10.1080/10826076.2012.743724.
- S. Fekete, L. Kizekai, Y.T. Sarisozen, N. Lawrence, S. Shiner, M. Lauber, Investigating the Secondary Interactions of Packing Materials for Size-Exclusion Chromatography of Therapeutic Proteins, J. Chromatogr.A, 1676 (2022) 463262.https://doi.org/10.1016/j.chroma.2022.463262.
- S. Fekete, A. Beck, J.L. Veuthey, D. Guillarme, Theory and Practice of Size Exclusion Chromatography for the Analysis of Protein Aggregates, J. Pharm.Biomed.Anal. 101 (2014) 161–173.http://dx.doi.org/10.1016/j.jpba.2014.04.011.
- R.D. Ricker, L.A. Sandoval, Fast, Reproducible Size-Exclusion Chromatography of Biological Macromolecules, J. Chromatogr.A 743 (1996) 43–50.https://doi.org/10.1016/0021-9673(96)00283-X.
- M. Kamberi, P. Chung, R. DeVas, L. Li, Z. Li, X. Ma, S. Fields, C.M. Riley, Analysis of Non-covalent Aggregation of Synthetic hPTH (1–34) By Size-Exclusion Chromatography and the Importance of Suppression of Non-specific Interactions for a Precise Quantitation, J. Chromatogr. B 810 (2004) 151–155.https://doi.org/10.1016/j.jchromb.2004.07.026.
- A.M. Striegel, W.W. Yau, J.J. Kirkland, D.D. Bly, Modern Size Exclusion Liquid Chromatography, John Wiley and Sons Inc., Hoboken, New Jersey, 2009.
- Cold Spring Harbour Laboratory Press (2006), Phosphate Buffered Saline (PBS), Recipe, https://doi.org/10.1101/pdb.rec8247.
ソリューション提供製品
720007790JA、2022 年 11 月