カラムのスケーリングとメソッドの最適化によるオリゴヌクレオチド精製の生産性の向上
研究目的のみに使用してください。診断用には使用できません。
要約
長年にわたり、低分子およびタンパク質ベースの医薬品が医薬品業界をリードしてきました。しかし、これらのモダリティで対処できる創薬の候補物質および治療の数には限りがあります。一方、特に遺伝情報の転写、スプライシング、翻訳が関わっている場合は、合成オリゴヌクレオチドを検討できる可能性があります。オリゴヌクレオチド医薬品は、20 ~ 30 mer の範囲の低分子合成核酸ポリマーのクラスで、遺伝子発現の調節に使用することができます。初期の探索段階から後期開発段階に至るまで、医薬品開発においては、さまざまな試験を円滑に行うのに十分な精製済み材料が必要になります。さらに、ポリメラーゼチェーン反応や定量ゲノミクスのアプローチでは、オリゴヌクレオチドを分子ツールとして使用します。この業界は新たなイノベーションにより拡大を続けており、精製スキームの改善も必要とされています。このアプリケーションノートでは、20 mer オリゴヌクレオチドの分析的分離と分取精製スキームの両方に用いられる、費用対効果が高いハイスループットの体系的アプローチを紹介します。
アプリケーションのメリット
- 分画後に簡単に除去できる、危険性が低く、より安価な移動相添加剤
- 分析的 UPLC 分離のワイドボア精製カラムへのスケーリング
- バッチ試験および選択した Oligonucleotide BEH™ C18 固定相を使用して、再現性のある分離および精製の性能を保証
- 効率の高い分離および精製のためのクロマトグラフィーメソッドパラメーターの最適化
- 分取オンカラムロードとフラクション回収の最適化による純度と回収率の最大化
はじめに
合成オリゴヌクレオチドの効率的な精製は、多くの場合困難な作業になります。現在まで、適切なカラムの選択、移動相の選択、グラジエント条件の最適化が、クロマトグラフィー担当者にとっての課題でした。オリゴヌクレオチドの分析および特性解析のための分離法の使用が、多数の文献で説明されています1–3。分析法は、高 pH および高温を使用して最適化される場合が多くなります。固定相の選択は、これらのクロマトグラフィーのニーズを考慮する必要があります。多くの科学者は、エチレン架橋型ハイブリッドシリカ(BEH)C18 吸着剤をこの目的のために選択しています。BEH C18 吸着剤カラムは、分析スケールおよび分取スケールのサイズで、XBridge C18 カラムとしてウォーターズから入手できます。オリゴヌクレオチドの分離を用いた BEH C18 粒子のバッチ検査および選択が、ACQUITY™ および XBridge™ Oligonucleotide BEH C18 の製造にさらに適用されています。
オリゴヌクレオチドの液体クロマトグラフィーは主に逆相イオン対(RP-IP)分離を用いて行われます。移動相にアルキルアミン添加剤を使用し、酢酸またはヘキサフルオロイソプロパノール(HFIP)を用いて緩衝化します。トリエチルアミン(TEA)/HFIP 水系移動相は、オリゴヌクレオチド LC-MS のゴールドスタンダードです。このタイプの移動相を使用することで、優れたピーク形状が得られます。また、酢酸の代わりに HFIP を使用すると、イオン化抑制が低減して、質量分析で溶出するオリゴヌクレオチドの検出がより容易になります。
オリゴヌクレオチド分離を分取スケールにスケーリングする際の課題の 1 つとして、HFIP は高価で危険性があることが挙げられます。ルーチンの分取スケールの分離に別のクロマトグラフィー条件を選択することで、HFIP への曝露を低減し、コストを削減できるというメリットがあります。第 2 に、粒子径の小さい吸着剤は、高圧が発生する可能性が高いため、通常は分取分離に適用できません。この問題に対する解決策として、流量を減らすこと、全多孔性の 2.5 µm の粒子を充塡したカラムを使用することが挙げられます。内径(ID)が 4.6 mm、10 mm、あるいは最大 50 mm のラボスケールの分取精製用カラムは、ウォーターズで市販されています。分取カラムに使用する最適ベッド密度(OBD™)充塡テクノロジーにより、分取に求められる機械的強度、再現性、効率に関連するカラム寿命が長くなり、ハイスループット精製が向上します1。
クロマトグラフィーの移動相を、HFIP ベースから酢酸トリエチルアンモニウム(TEAA)または酢酸ヘキシルアンモニウム(HAA)をベースにした移動相に置き換えることができます2。 TEAA と HAA はいずれも揮発性であり、蒸発乾固または凍結乾燥によって、回収したオリゴヌクレオチドフラクションから除去することができます。炭素数が多いさまざまな代替のアルキルアミンを、オリゴヌクレオチドの分離用のイオン対試薬として使用できることが示されています2。 アルキルアミンの選択は、精製するオリゴの特性に影響されます。しかし、炭素長が増加すると、通常は揮発性が低下し、蒸発乾固や凍結乾燥にかかる時間とエネルギーコストの両方が増大します。揮発性の移動相および添加剤を用いるメソッドを使用することは、コストの削減、ラボの安全性確保、精製または精製後のステップ数低減の上で非常に重要です。
このアプリケーションノートでは、TEA/HFIP 移動相に基づく分析スケールの UPLC™ 分離から、HFIP を使用しないワイドボア HPLC 精製法までの、オリゴヌクレオチド精製法のスケールアップについて実証します。
実験方法
20 mer オリゴヌクレオチドモデル混合物の LC 分析
サンプル前処理
20 mer(5'-G-C-C-T-C-A-G-T-C-T-G-C-T-C-C-A-C-C-T-3)の粗体を、この試験のプローブオリゴヌクレオチド化合物として使用するために、Oligo Factory(マサチューセッツ州ホリストン)から購入しました。サンプル溶媒は、粗オリゴヌクレオチド混合物を 10 mM NH4OAc に再溶解して、合計濃度 10 µM(59 µg/mL)になるように調製しました。
移動相の調製
8.6/100 mM TEA/HFIP 混合液を移動相 A として調製しました。メタノールを移動相 B として使用しました。その他の実験では、100 mM TEAA(pH 7.0)を調製して移動相 A として、アセトニトリルを移動相 B として使用しました。
カラム
この試験では、XBridge Oligonucleotide BEH C18 130 Å 2.5 µm カラムを使用します。これらのカラムには、オリゴヌクレオチド分離性能試験用に特別にテストし、ロットを選択した BEH C18 固定相のバッチが入っています。米国マサチューセッツ州トーントンで実施した QC テストでは、イオン対条件下でシャープで対称的なピークが得られる固定相のバッチのみが、オリゴヌクレオチド分析カラム用に割り当てられています。この点は、最も再現性の高いメソッドおよび精製手法を実現して維持することを目指す際に、重要な検討事項となります。
初期分析メソッド
|
移動相 A: |
8.6/100 mM TEA/HFIP |
|
移動相 B: |
MeOH |
|
カラム: |
ACQUITY UPLC Oligonucleotide BEH C18 2.1 × 100 mm、1.7 µm(製品番号:186003950) |
|
流速: |
0.4 mL/分グラジエント |
|
冷却温度: |
60 ℃ |
|
希釈溶媒: |
10 mM NH4OAc 水溶液 |
|
サンプル濃度: |
10 µM(59 µg/mL) |
|
注入量: |
2 µL(120 ng オンカラム) |
|
グラジエント: |
5% B で 0.25 分間、10 分間で 5 ~ 15%、0.25 分間で 15 ~ 95%、95% で 1 分間ホールド、0.1 分間で 95 ~ 5%、5% で 3.4 分間ホールド。 |
最適化した分析スケールの TEAA を用いるメソッド
|
移動相 A: |
100 mM TEAA、pH 7.0 |
|
移動相 B: |
アセトニトリル |
|
カラム: |
XBridge Oligonucleotide BEH C18 130 Å 2.5 µm 4.6 × 50 mm(製品番号:186003953) |
|
流速: |
0.8 mL/分グラジエント |
|
カラム温度: |
25 ℃ |
|
希釈溶媒: |
100 mM TEAA、pH 7.0 |
|
サンプル濃度: |
1 mg/mL |
|
注入量: |
10 µL(10 µg オンカラム) |
|
グラジエント: |
0.5 分間で 1 ~ 7.8%、10 分間で 7.8 ~ 9.8%、0.1 分間で 9.8 ~ 19%、1.4 分間 19% でホールド、0.1 分間で 19 ~ 1%、4.9 分間 1% でホールド |
最適化した分取スケールの TEAA を用いるメソッド
|
移動相 A: |
100 mM TEAA、pH 7.0 |
|
移動相 B: |
アセトニトリル |
|
カラム: |
XBridge Oligonucleotide BEH C18 130 Å OBD Prep 30 × 50 mm 2.5 µm(製品番号:186008963) |
|
流速: |
25 mL/分のグラジエント 40 mL/分のクリーンアウトおよび平衡化 |
|
カラム温度: |
25 ℃ |
|
希釈溶媒: |
100 mM TEAA、pH 7.0 |
|
サンプル濃度: |
10 mg/mL |
|
注入量: |
42 µL、その後変更 |
|
グラジエント: |
0.2 分間で 1 ~ 7.8%、12 分間で 7.8 ~ 9.8%、0.1 分間で 9.8 ~ 19%、0.9 分間 19% でホールド、0.1 分間で 19 ~ 1%、2 分間 1% でホールド |
結果および考察
分析スケールでの HFIP からの切り替え
20 mer オリゴヌクレオチドの分離はまず、ACQUITY UPLC Oligonucleotide BEH C18 1.7 µm 2.1 × 100 mm カラムを使用して分析スケールで行いました。このメソッドでは、移動相の一部として HFIP を使用し、カラム温度を 60 ℃ に設定しました。図 1 に、この条件で得られたクロマトグラムの例を示します。二次構造の影響を排除し、オリゴヌクレオチドの拡散を高めるために、カラム温度 60 ℃ を使用しました。ほとんどの短いオリゴヌクレオチドは二次構造がないため、室温で十分に分離できるので、分取スケールへのスケールアップが大幅に簡素化します。
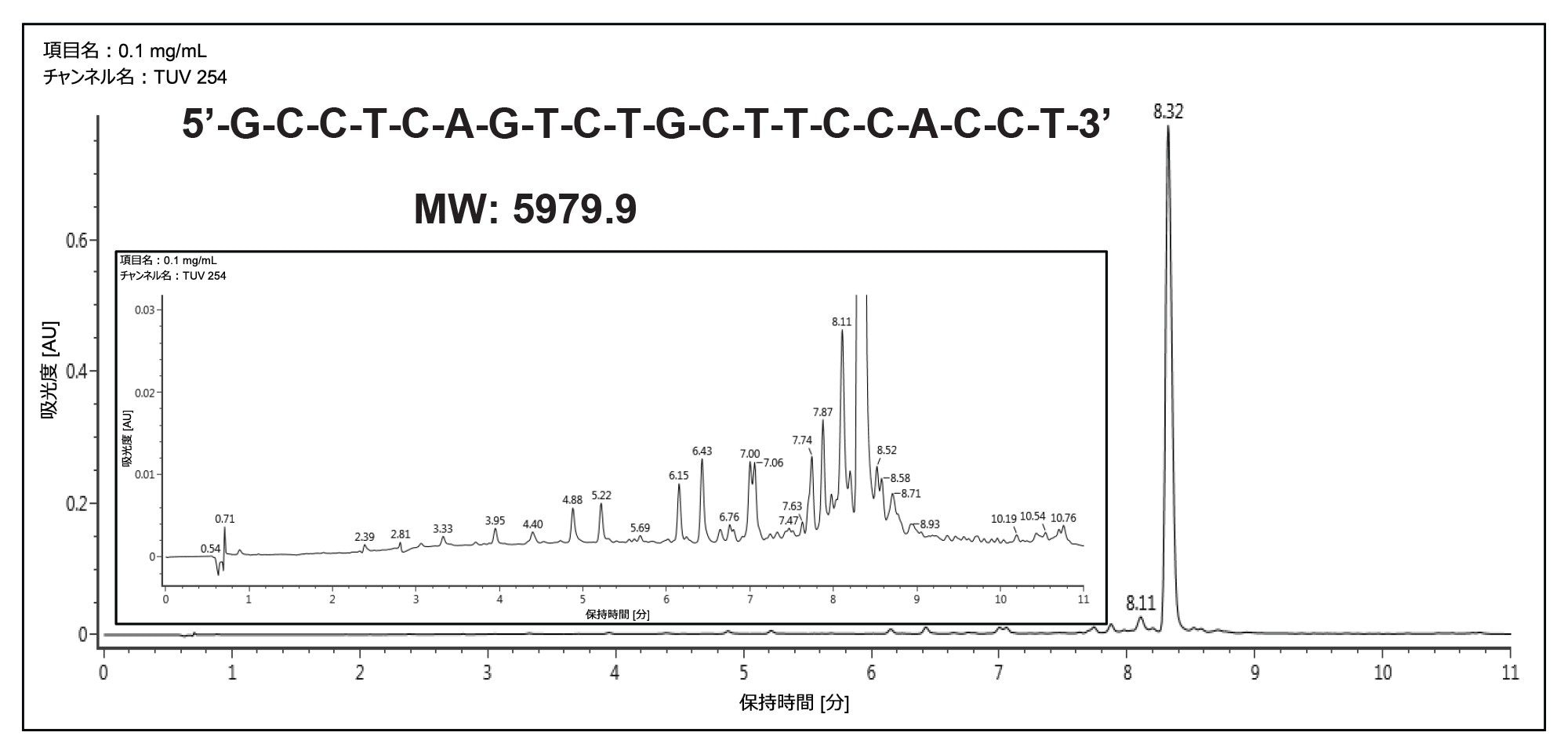 図 1. カラム温度 60 ℃ で、HFIP-TEA 移動相および ACQUITY UPLC Oligonucleotide BEH C18 130 Å 1.7 µm 2.1 × 100 mm カラムを使用した 20 mer オリゴヌクレオチドの分析的分離。ターゲット分子種の純度は約 90% と算出されます。
図 1. カラム温度 60 ℃ で、HFIP-TEA 移動相および ACQUITY UPLC Oligonucleotide BEH C18 130 Å 1.7 µm 2.1 × 100 mm カラムを使用した 20 mer オリゴヌクレオチドの分析的分離。ターゲット分子種の純度は約 90% と算出されます。
次の実験では、酢酸アンモニウム(TEAA)を使用するメソッドを分析スケールで開発しました。このメソッドを、4.6 × 50 mm XBridge™ Oligonucleotide BEH C18 2.5 µm カラム(100 mM TEAA、pH 7.0 で構成される移動相 A およびアセトニトリルの移動相 B)を使用して実行しました。カラムを 25 ℃ に維持し、出発組成 5% B のマルチステップグラジエントでオリゴヌクレオチドを溶出させました。次に、組成を 5% B から 8% B まで 0.25 分で増加させ、続いて 8% B から 12% B まで 9.75 分で増加させた後、最終的に 12% B から 95% B まで 0.1 分間で増加させました。さらに 0.9 分間 95% B でホールドしてから 0.1 分間で 5% B に戻しました。カラムを 4.9 分間 5% B で平衡化させてから次の注入を行いました。図 2 に、得られたクロマトグラムを示します。TEAA での n-1 と n-x の分離は、最初の TEA/HFIP での実行ほど効率的ではありませんが、この分離でも、反応終了した失敗配列からターゲットのピークを分離することができました。このメソッドにより、後続のフラクション回収および乾燥のステップにおける溶媒の揮発性を維持しつつ、費用対効果が高く危険性の低い溶媒システムが実現します。
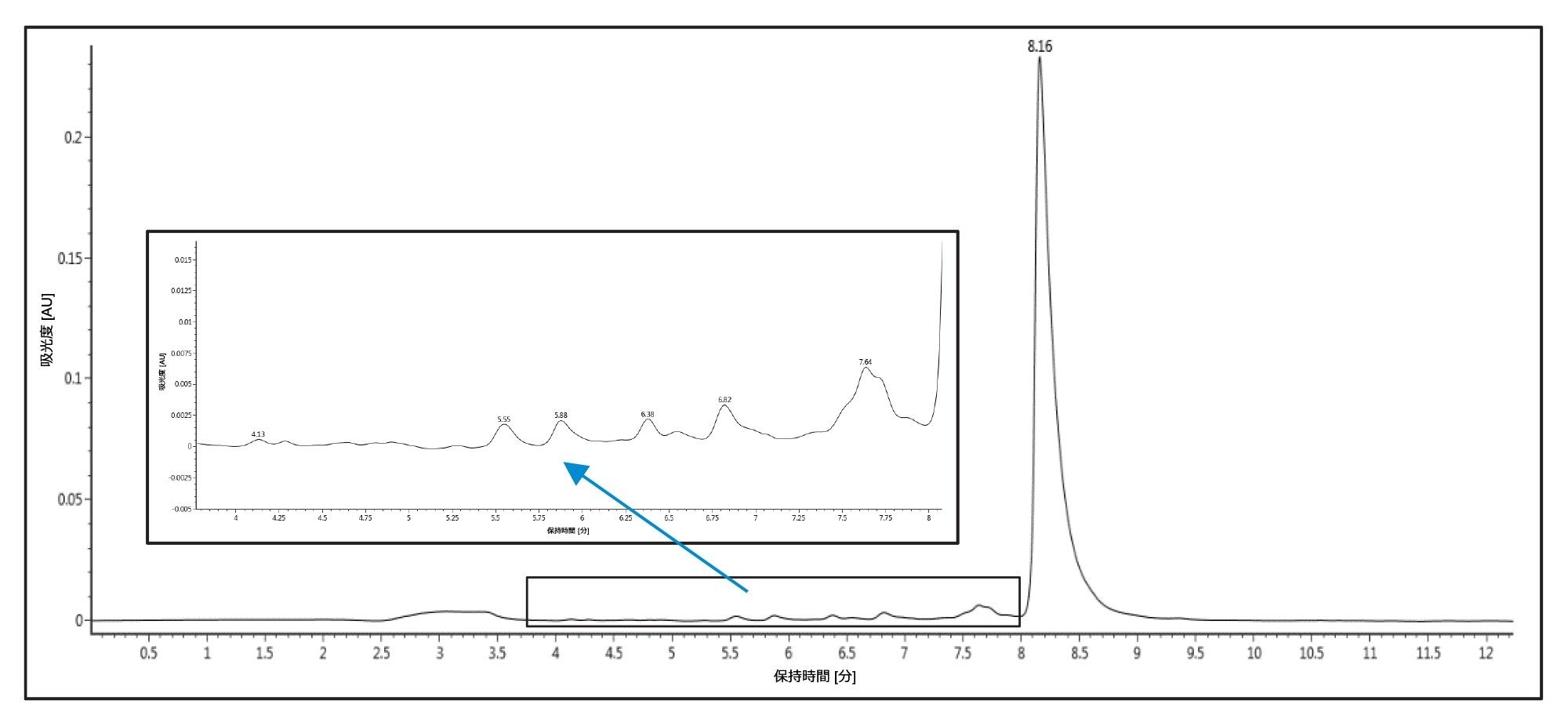 図 2. カラム温度 25 ℃ で、TEAA 移動相および XBridge Oligonucleotide BEH C18 130 Å 2.5 µm 4.6 × 50 mm カラムを使用した 20 mer オリゴヌクレオチドの分析的 HPLC 分離
図 2. カラム温度 25 ℃ で、TEAA 移動相および XBridge Oligonucleotide BEH C18 130 Å 2.5 µm 4.6 × 50 mm カラムを使用した 20 mer オリゴヌクレオチドの分析的 HPLC 分離
分取用ワイドボアカラムへのスケーリング
次に、Waters™ グラジエントカリキュレーターを使用して、TEAA を使用するこの新しいメソッドを、30 × 50 mm XBridge Oligonucleotide BEH C18 OBD Prep 2.5 µm カラムおよび 3767 オートサンプラー/フラクションコレクター、SFO、UV 検出器(PDA または TUV)、SQD2 シングル四重極 MS、MS 用の ACQUITY ISM メイクアップフローポンプで構成されるウォーターズの自動精製システムに移行しました。この研究のこの部分では、ピーク全体にわたる純度を評価することが目標であったため、時間ベースの回収を使用しました。一方、MS ベースのフラクション回収を使用することも可能で、ターゲットの質量のみを回収することにより通常は選択性を向上させることができます。内径が異なるカラムの間でメソッドをスケーリングする場合、考慮すべき最も重要なパラメーターの 1 つは、カラム半径の 2 乗に比例するカラム流速です。さらに、内径(ID)4.6 mm のカラムから内径 30 mm のカラムにスケールアップする場合、カラム容量の比に基づいてスケーリングしたロード係数は 42 倍になります。したがって、分析注入 10 µg は分取注入 410 µg に変換されます。この質量ロードで得られた分取スケールのクロマトグラムを図 3 に示します。フラクション回収を時間でトリガーし、1 チューブあたり 15 秒間の溶出液を回収しました。最初の UPLC メソッドを使用して、回収した個々のフラクションとプールしたフラクションを分析すると、個々のフラクションの純度は 3 つの主要フラクション(図 3 で F1 ~ F3 と表記)にわたって約 98% ~ 80% の範囲であり、プールしたフラクションの純度は 96.3% でした。これらのフラクションの分析的純度分析を図 4 に示します。プールしたフラクションの全体的な回収率は 85% と算出されました。純度を高める必要がある場合は、3 番目のフラクションをプールしないことが推奨されます。F1 と F2 をプールしたフラクションの純度は 97% を超えており、回収率に与えた影響は約 5% と考えられます。
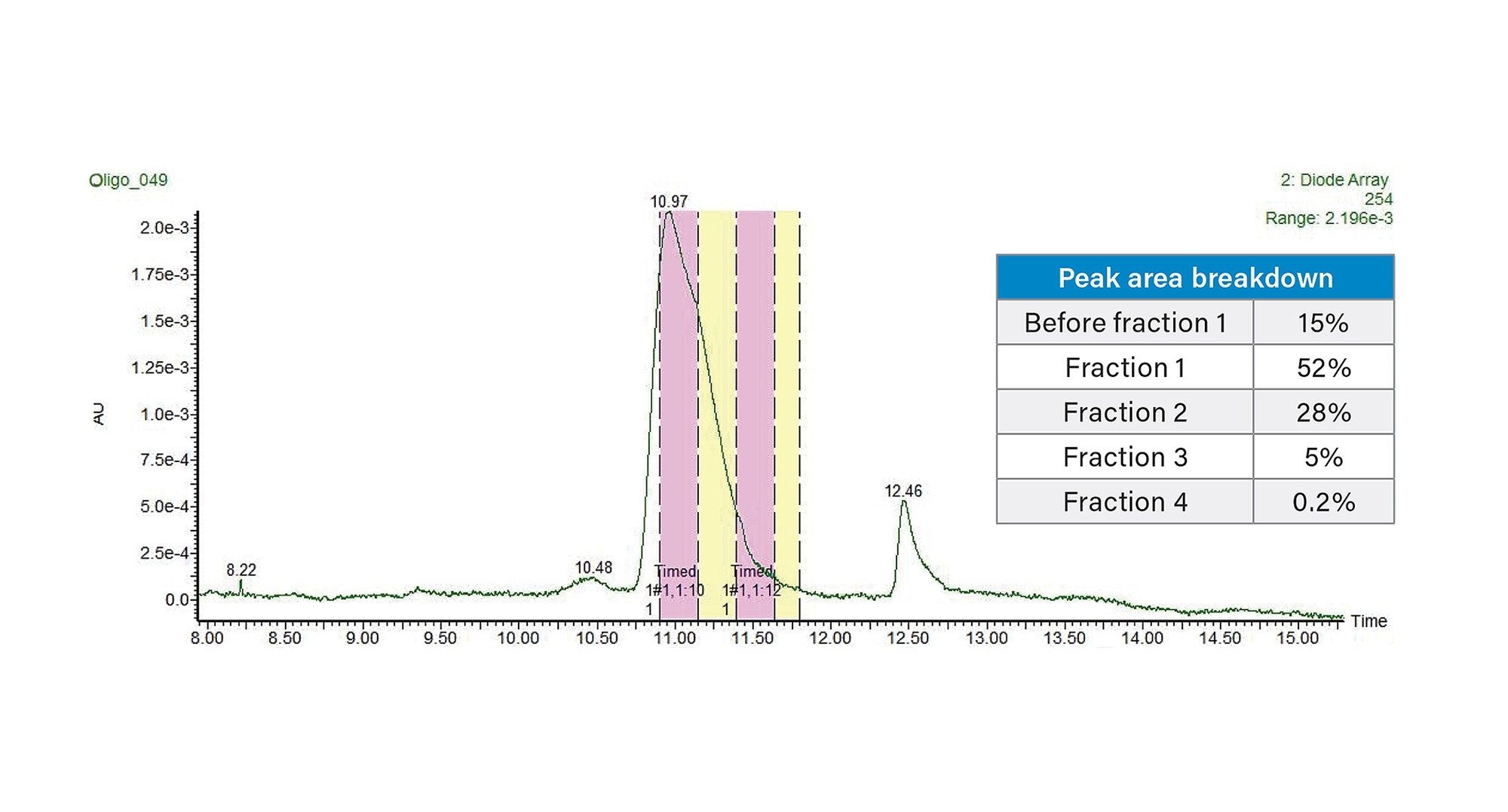 図 3. TEAA 移動相、質量ロード 410 µg、周囲カラム温度、30 × 50 mm XBridge Oligonucleotide BEH C18 OBD Prep 2.5 µm カラムを使用して、20 mer オリゴヌクレオチドについて得られた分取クロマトグラム
図 3. TEAA 移動相、質量ロード 410 µg、周囲カラム温度、30 × 50 mm XBridge Oligonucleotide BEH C18 OBD Prep 2.5 µm カラムを使用して、20 mer オリゴヌクレオチドについて得られた分取クロマトグラム
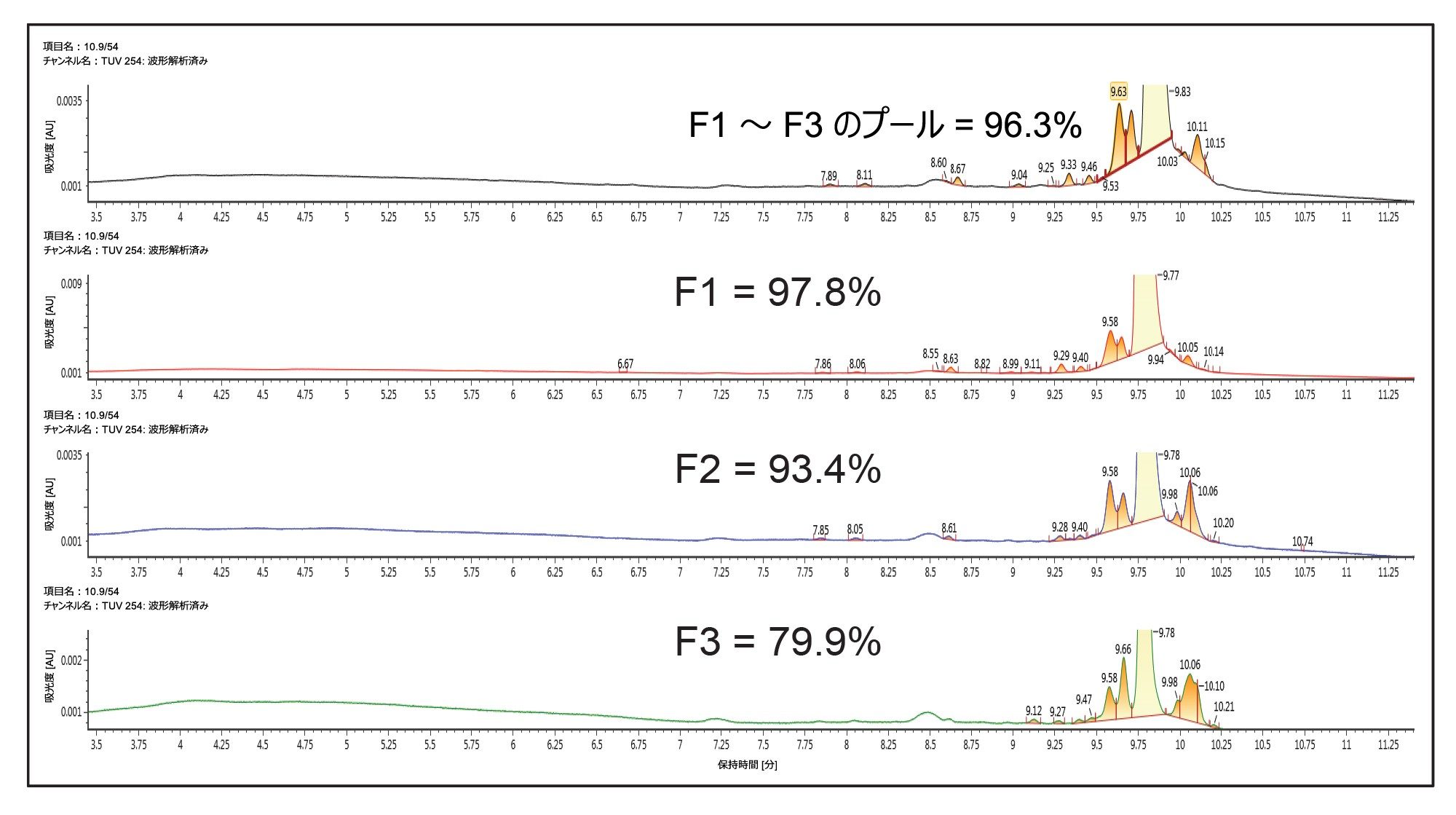 図 4. カラム温度 60 ℃ で、HFIP–TEA 移動相および ACQUITY UPLC Oligonucleotide BEH C18 130 Å 1.7 µm 2.1 × 100 mm カラムを使用した、図 3 のクロマトグラムに対応するフラクションの UPLC による純度分析。
図 4. カラム温度 60 ℃ で、HFIP–TEA 移動相および ACQUITY UPLC Oligonucleotide BEH C18 130 Å 1.7 µm 2.1 × 100 mm カラムを使用した、図 3 のクロマトグラムに対応するフラクションの UPLC による純度分析。
質量ロードの増加
図 3 および 4 に示した精製結果は、頑健な分析的分離の性能を失うことなく、性能を失うことなく 30 mm の分取カラムにスケーリングできることを実証しています。一方、精製メソッドの成功の別の尺度として、生産性が挙げられます。このメソッドにより、1 時間あたり約 1 mg の精製オリゴヌクレオチドが得られています。生産性を改善するための 1 つの選択肢として、各注入の質量ロードを増やすことが挙げられます。このアプローチの課題は、ロード量に依存してピークが広がるため、精製済み物質を得るための分離度や回収率が低下する可能性があることです。図 5 に、質量ロードを 25 mg から最大 50 倍増やした場合に、30 × 50 mm 2.5 µm オリゴヌクレオチド RPLC カラムでのクロマトグラフィーに出る影響を示しています。予想どおり、ロード量を増やすにつれて、ピークの立ち上がりがさらに前に移動しました。
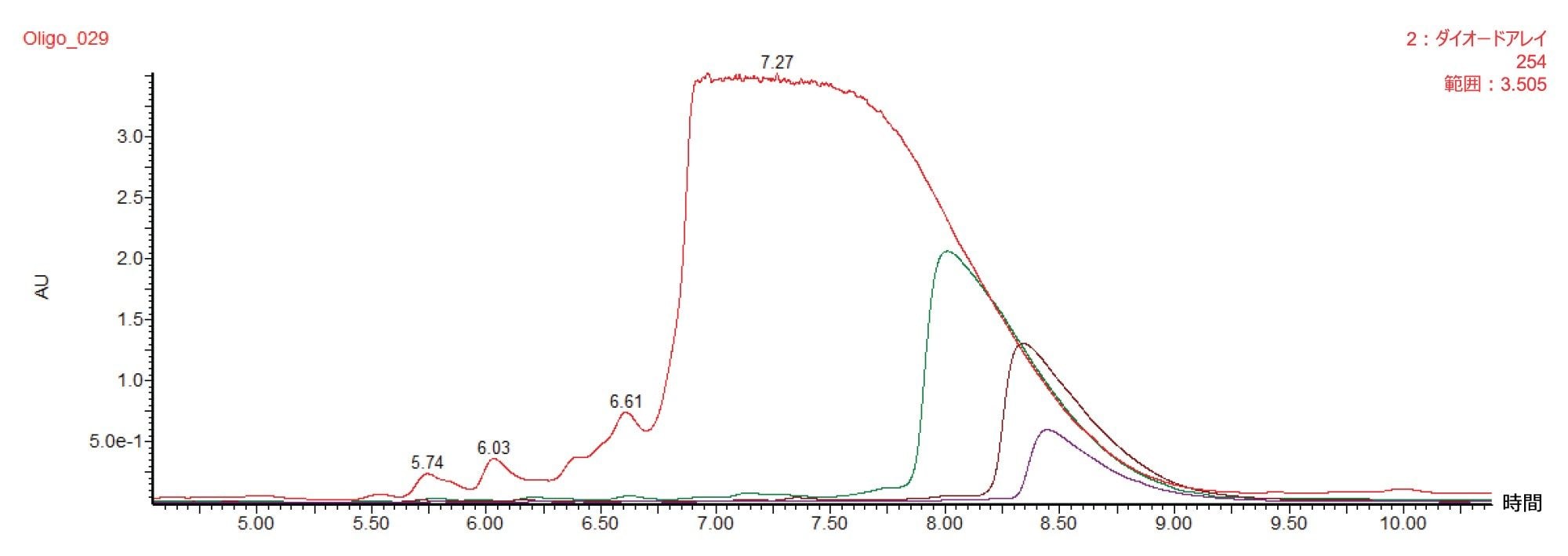 図 5. 30 × 50 mm XBridge Oligonucleotide BEH C18 OBD Prep 2.5 µm カラムで質量ロードを増やして得られた 20 mer オリゴヌクレオチドの分取クロマトグラムの重ね描き
図 5. 30 × 50 mm XBridge Oligonucleotide BEH C18 OBD Prep 2.5 µm カラムで質量ロードを増やして得られた 20 mer オリゴヌクレオチドの分取クロマトグラムの重ね描き
ピーク全体の純度の評価
図 5 に示した各注入では、15 秒間隔でフラクションを回収し、UPLC HFIP/TEA メソッドによって濃縮および分析を行いました。これらの純度測定は、図 6 および 7 に示しています。以前に報告されているように、立ち上がりから頂点までのピーク領域は、ピークの他の領域に比べて純度がかなり低くなっています3。 一方、質量ロードの増大とともにピークが広がるため、純粋なフラクションの数も増加し、結果として回収率と生産性が向上しています。全体的な純度要件に基づいて必要なチューブをプールし、最終的な精製物質を得ることができます。表 1 に、質量ロードと異なる分取アプローチによって得られる生産性の計算を示します。
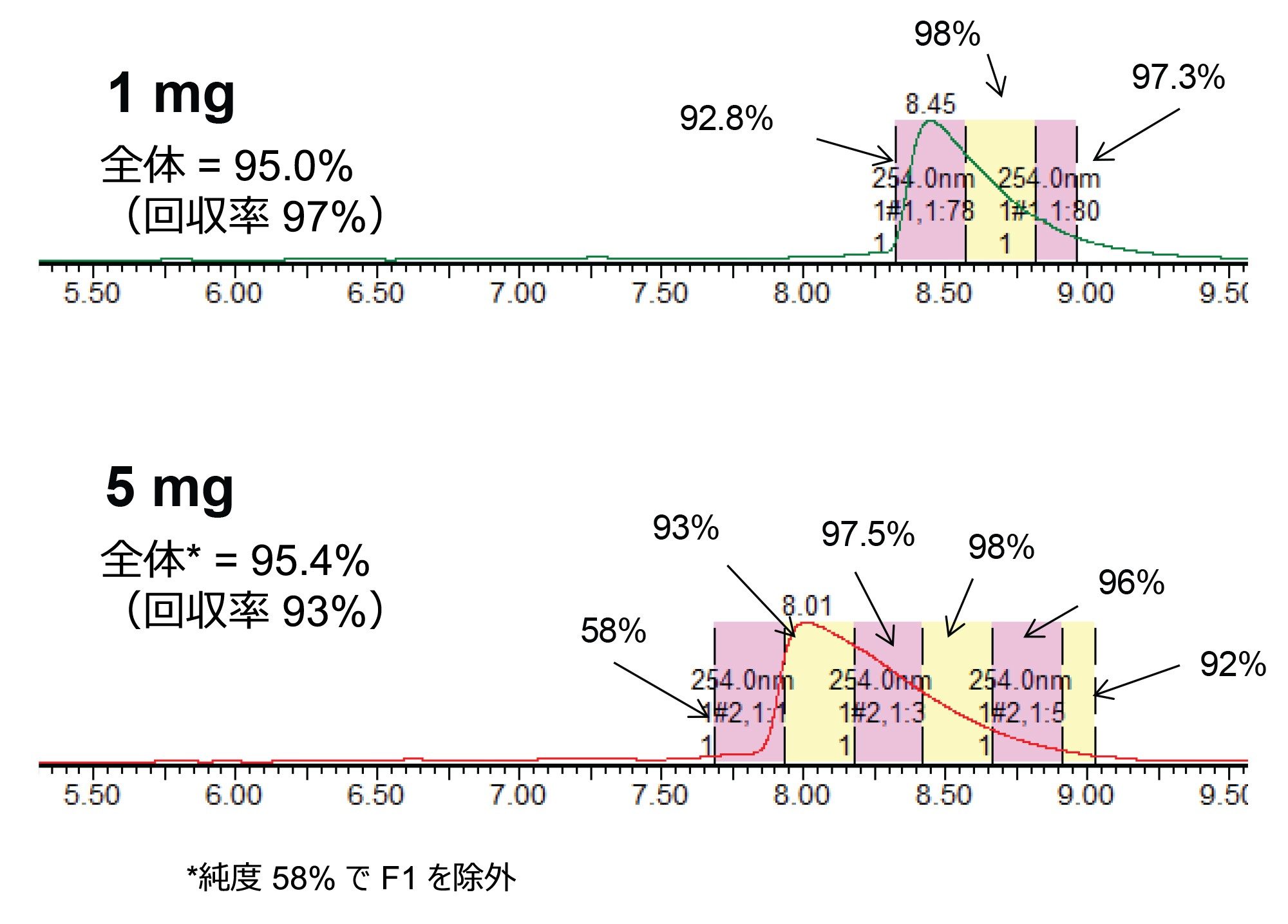 図 6. 1 mg および 5 mg の精製におけるフラクションの純度の測定
図 6. 1 mg および 5 mg の精製におけるフラクションの純度の測定
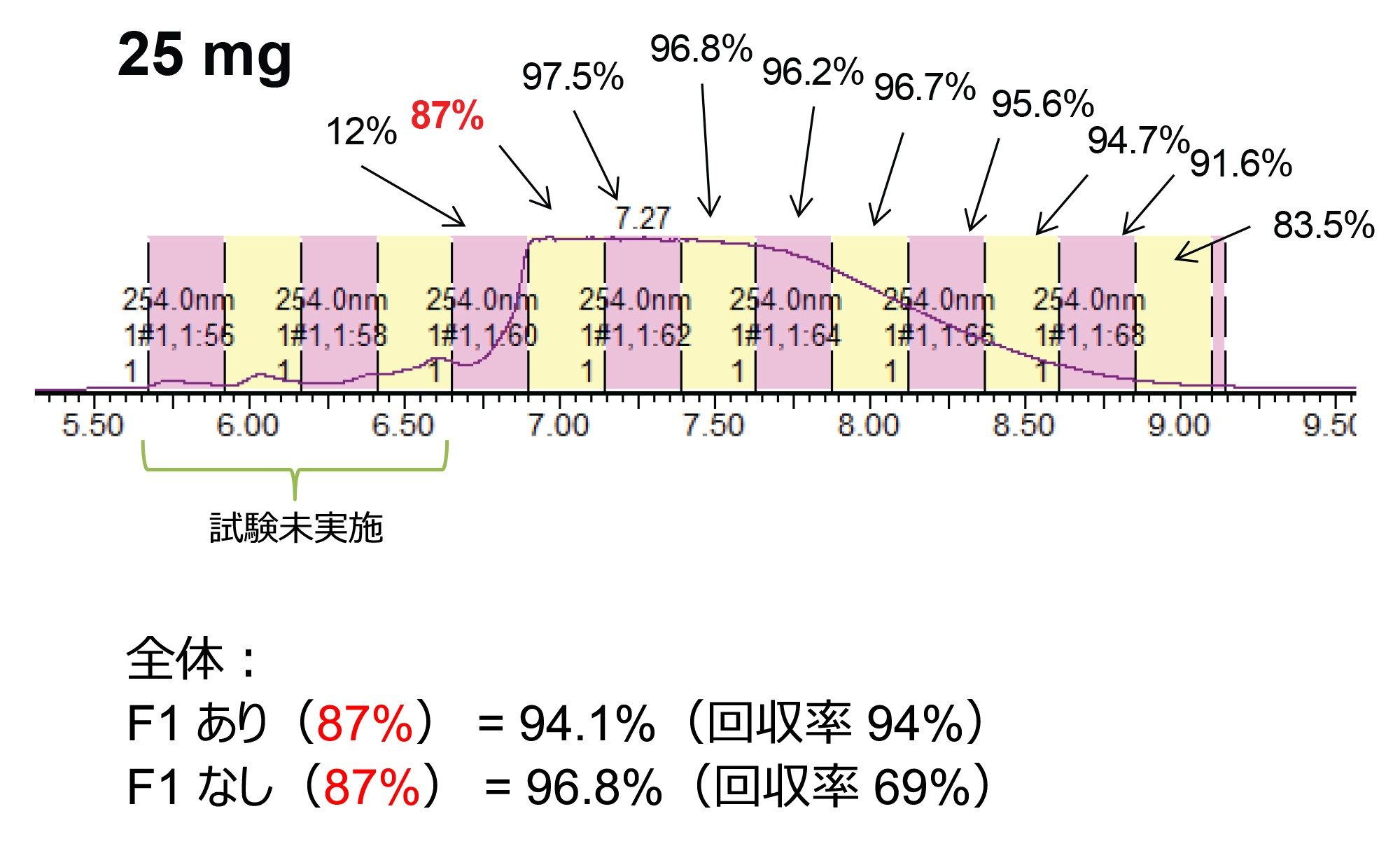 図 7. 25 mg の精製におけるフラクションの純度の測定
図 7. 25 mg の精製におけるフラクションの純度の測定
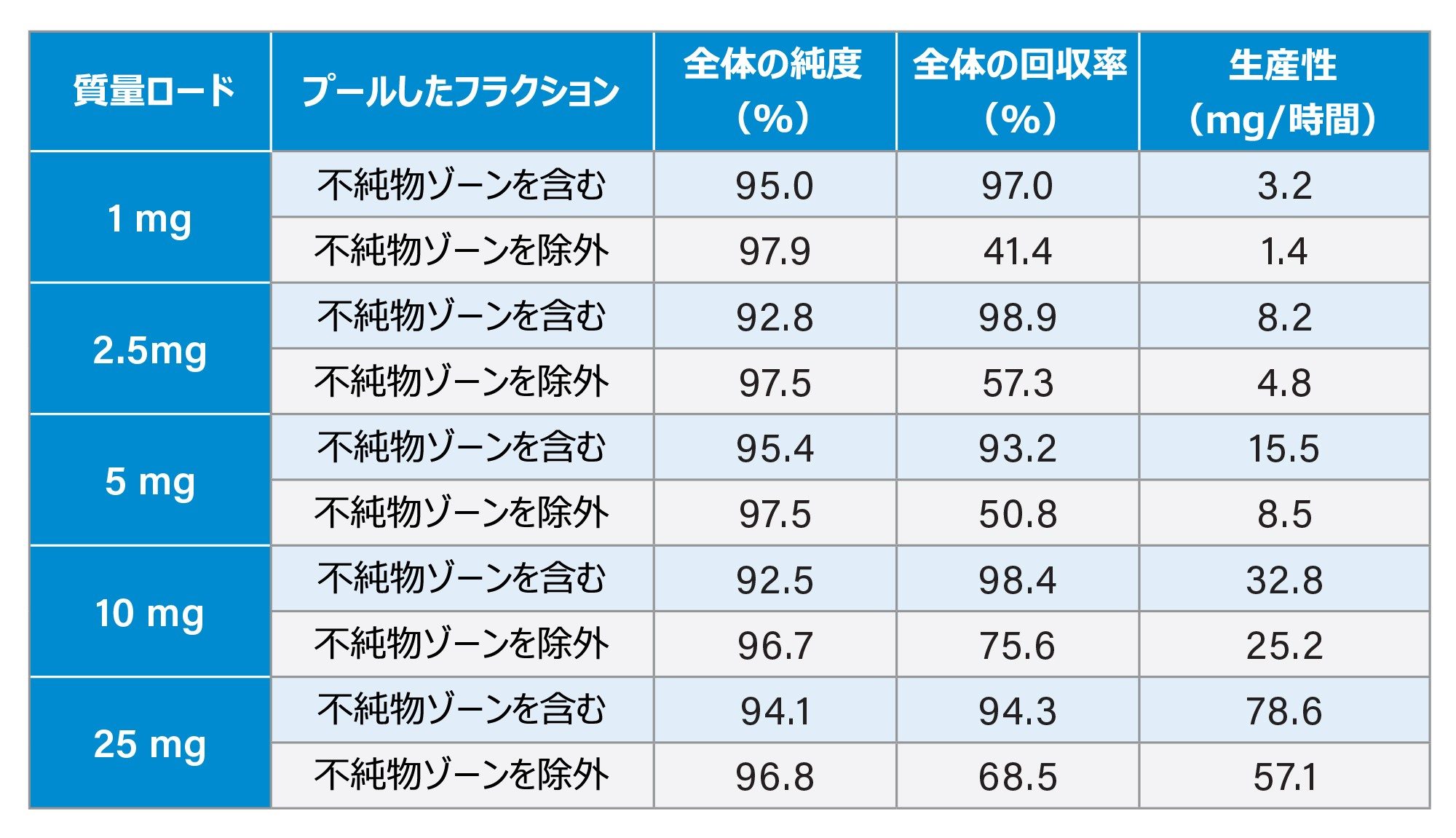 表 1. 様々な質量ロードでの純度、回収率、および得られた生産性
表 1. 様々な質量ロードでの純度、回収率、および得られた生産性
メソッドパラメーター
生産性に影響する変数への理解を深めるために、精製条件についてさらに検討しました。流速が低いと分離は改善しますが、生産性が低下します。グラジエント流速を 17 mL/分から 25 mL/分に増加させることについて評価しました。表 2 から、純度はわずかに低下しましたが、生産性が 50% 向上していることがわかります。
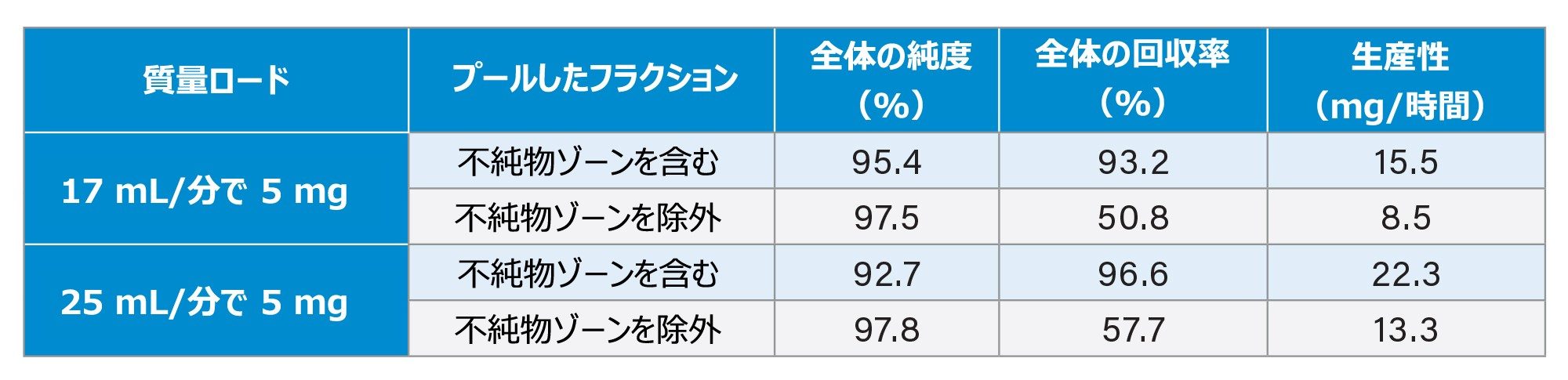 表 2. さまざまなグラジエント流速での純度、回収率、および得られた生産性
表 2. さまざまなグラジエント流速での純度、回収率、および得られた生産性
生産性を最適化するための別のアプローチとして、フォーカスグラジエントを用いる方法があります。前の例では、フォーカスグラジエントをすでに用いていたため、溶出ウィンドウの外側における流速の増加を調査しました。再生および再平衡化のステップでの移動相流速を増加させると、時間を大幅に節約することができました。
また、オートサンプラーが洗浄と次のサンプル注入を行っている間に、その時間をカラムの平衡化に使用することで、メソッドの実行時間がわずかに短縮しました。この例では、実行時間が 1 分短縮しました。これと流速の変更を合わせて、全体的な生産性が約 25% 向上しました4。 最適化した分取クロマトグラフィーメソッドを表 2 に示します。これにより、必要な純度に基づいて、精製オリゴヌクレオチドを約 75 ~ 100 mg/時間の速度で生成できるメソッドが得られます。
最後に、他の寸法のカラム(特にボアがさらに大きいカラム)の使用を検討する必要があります。これにより生産性がさらに向上しますが、今回は試験していません。カラムの内径を 30 mm から 50 mm に増やすと、ローディングキャパシティと生産性が約 3 倍増加しますが、流速 47.2 mL/分を用いる必要があります。オフラインのカラムクリーニングおよび平衡化を導入することによって、生産性をさらに高めることができます4。 これには、切り替えバルブ、再生ポンプ、2 本目のカラムなどの追加のハードウェアが必要です。これによって、この時間がメソッドで完全に不要になります。
結論
このアプリケーションノートでは、効率の高い分析的純度測定と生産性の高い精製メソッドの間の関係性および拡張性について説明しています。HFIP 移動相は LC-MS による分析的純度測定には適していますが、精製プロセスに使用すると問題が生じます。そのため、HFIP 移動相での分析的分離を、ワイドボア最適ベッド密度分取カラムでの TEAA ベースの精製に正常に移行できることを実証しました。メソッドパラメーターと質量ロードの最適化により、100 mg/時を超える速度で純度 95% を超える物質を生成できる、シンプルなラボスケールの精製スキームを考案しました。分取カラムの内径を大きくし、オフラインでのカラムのクリーニングと再生を導入することで、さらなる加速が可能になります。これらの手法は、拡張を続ける合成オリゴヌクレオチド業界をサポートする新たなイニシアチブに対応しており、ウォーターズでは、これが業界にとって価値の高い手法になることを願っています。
参考文献
- Patrik D. McDonald et al., Optimum Bed Density [OBD™] columns: Enabling Technology for Laboratory-Scale Isolation and Purification 720001939.
- Gilar, M. et al., Best Practices for Oligonucleotide Analysis Using Ion-Pair Reversed - Phase Liquid Chromatography -Columns and Chemistries 720006948.
- Gilar, M. et al., HPLC and UPLC column for Analysis of Oligonucleotides, Waters 720002376.
- Lefebvre, P. et al., Evaluating the Tools for Improving Purification Productivity.Waters application note 720001696.2007
720008266JA、2024 年 2 月