Many of the benzodiazepines are potentially addictive1 and longterm use can lead to dependency. Consequently their analysis is of key importance in both clinical and forensic settings. Misuse of these medications by vulnerable populations such as the elderly2 or the mentally-ill3 is common. The elderly are at particular risk, as sensitivity to benzodiazepines tends to increase with age thus, these analytes are commonly reported in self-poisonings4-7. Recreational use of benzodiazepines has also been reported; they are often used in combination with other narcotics e.g., they can be used to augment the ‘high’ of heroin or cocaine or can be used to reduce the after-effects of LSD or amphetamine use8. Drug-facilitated crime often involves benzodiazepines due to their sedative properties and amnesia-producing effects9.
Traditional techniques used for the quantitation of benzodiazepines include gas chromatography/mass spectrometry (GC-MS) and high performance liquid chromatography/mass spectrometry (HPLC-MS). The former typically requires inclusion of an additional derivatisation step and analysis can be problematic due to the thermo-labile nature of some of the analytes within this class. In contrast, HPLC-MS can separate a wide range of substances without the need for derivatisation and utilises ‘softer’ ionisation techniques e.g., electrospray ionisation (ESI) which allows the protonated molecular species to remain intact.
Since its introduction in 2004, UltraPerformance LC (UPLC) has repeatedly demonstrated significant advantages compared to HPLC-based methods e.g., enhanced resolution, sensitivity and throughput. Thus our aim was to develop a method utilising this technique.
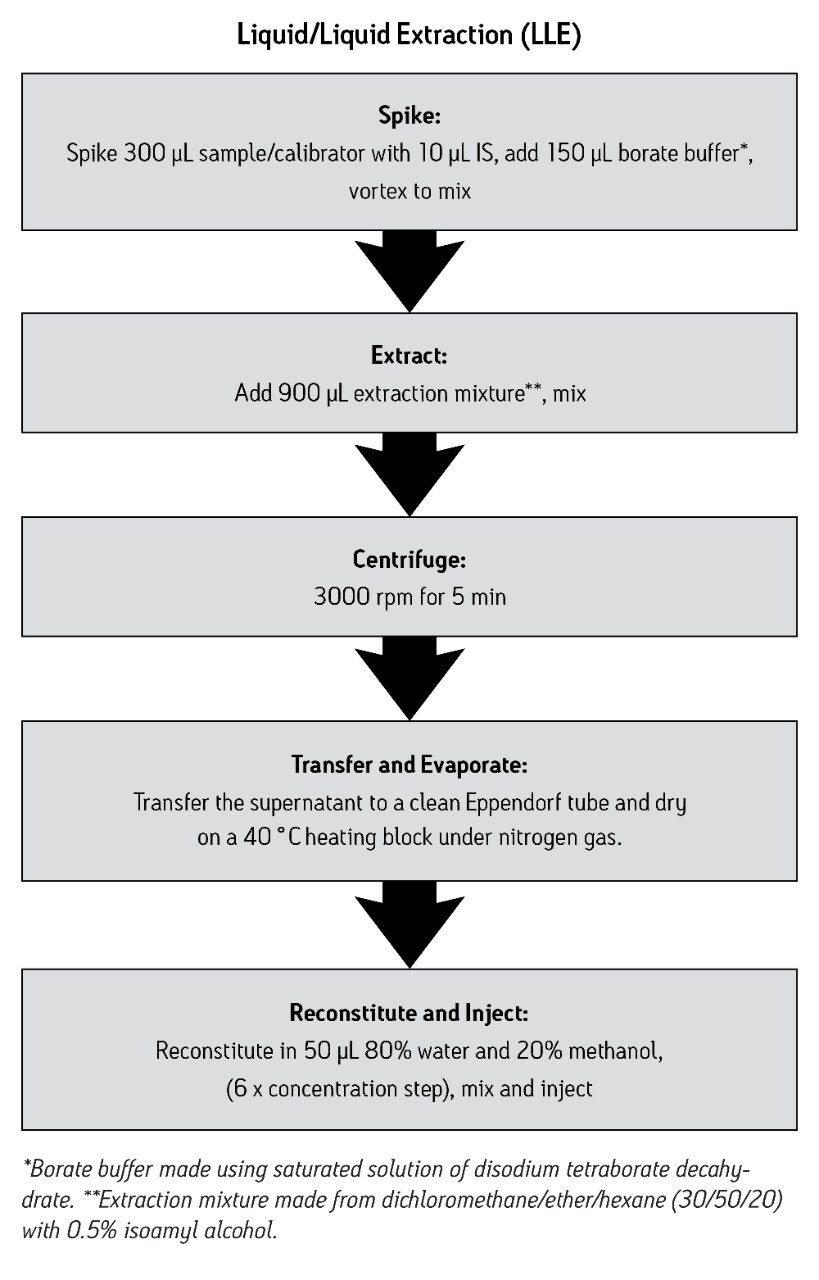
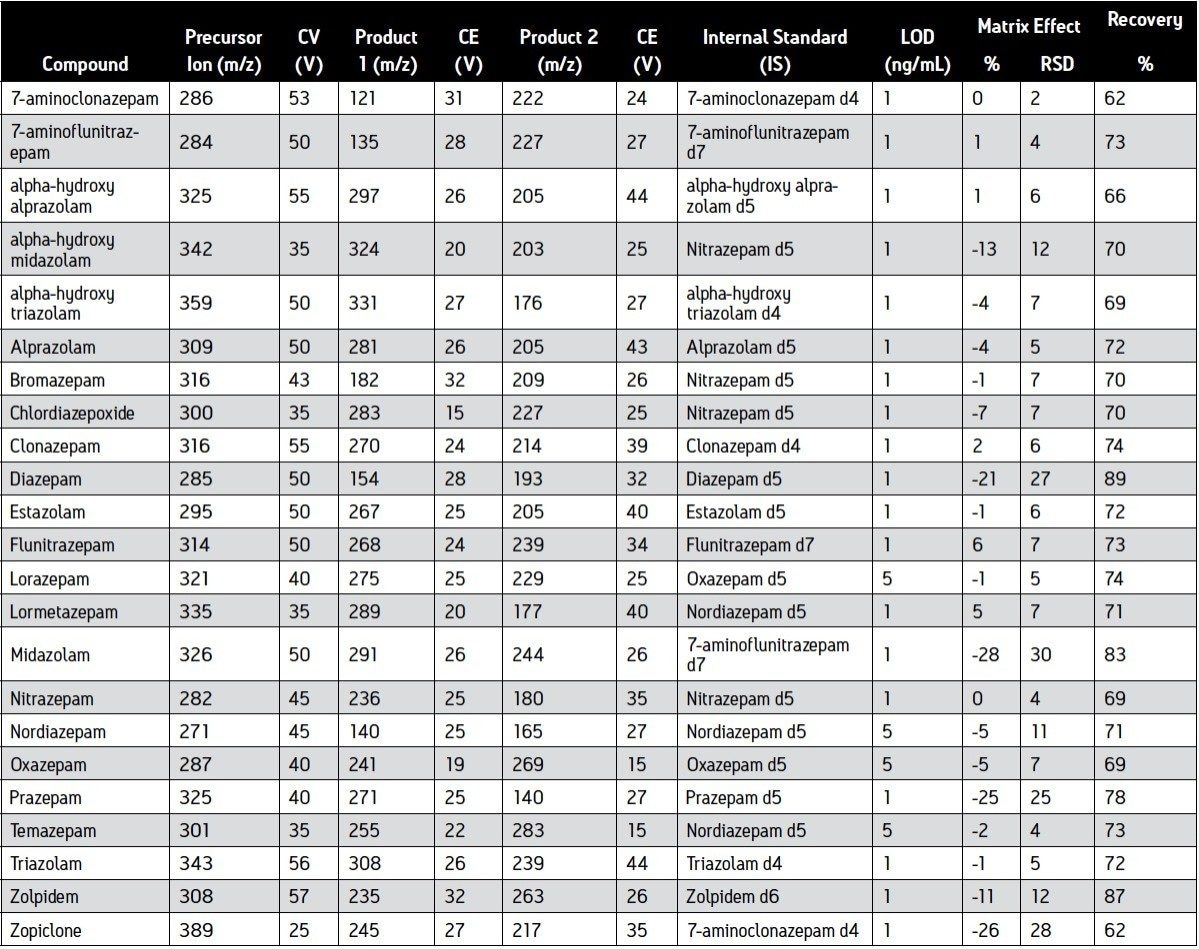
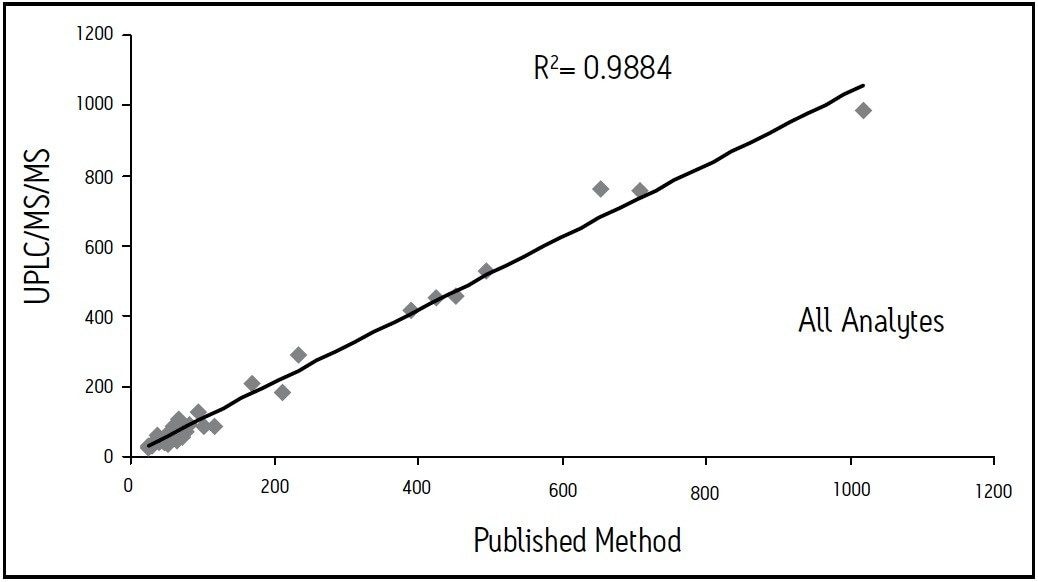
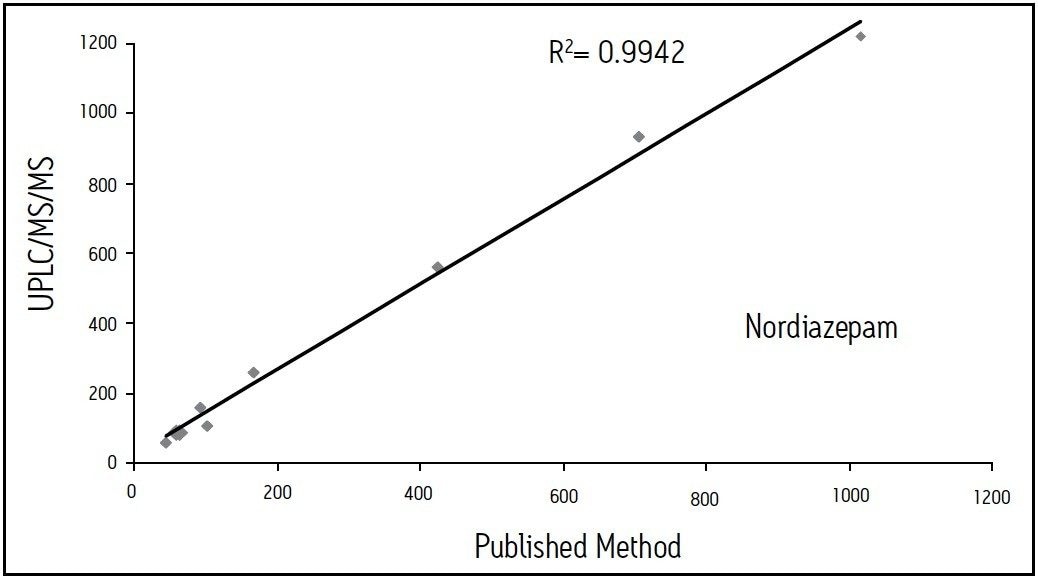
We describe a quantitative method based on liquid/liquid extraction (LLE) and UPLC with tandem mass spectrometry (MS/MS). The method’s performance has been evaluated using authentic samples. Data were compared to results obtained with a validated method based on HPLC-MS/MS10.