Mycotoxins are toxic secondary metabolites produced by various mold species that grow on many agricultural commodities and processed food, either in the field or during storage.1,2 They have been ranked as the most important chronic dietary risk factor, higher than synthetic contaminants, plant toxins, food additives, or pesticide residues.3
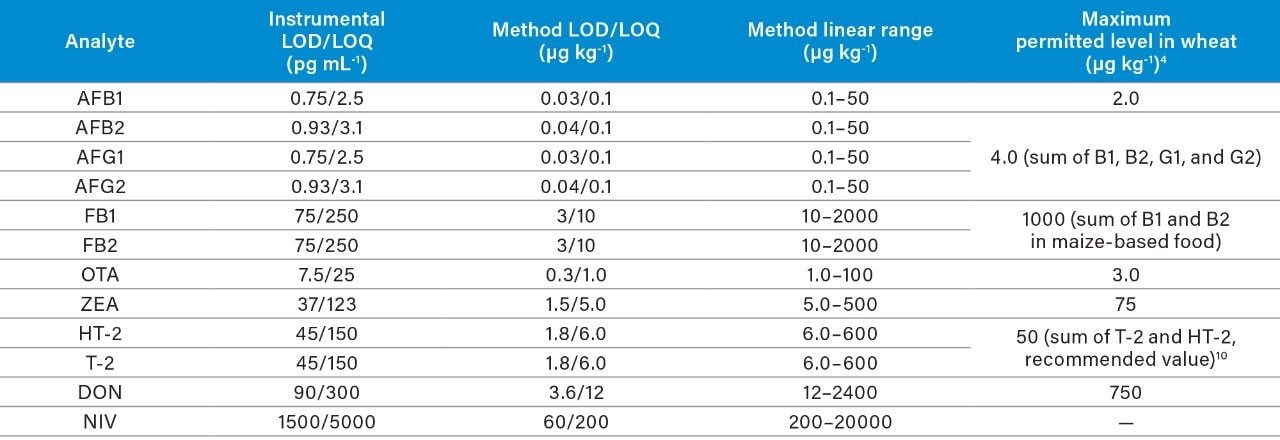
The European Commission Regulation No. 1881/2006 (and subsequent amendments)4 set the Maximum Permitted Levels (MPLs) for aflatoxins B1, B2, G1 and G2, fumonisins B1 and B2, deoxynivalenol, toxins T-2 and HT-2, zearalenone and ochratoxin A in different foodstuffs. The most recent EFSA scientific opinion established a Tolerable Daily Intake (TDI) of 1.2 μg kg-1 body weight per day for nivalenol,5 whose highest mean concentrations are typically observed in oats, maize, barley, and wheat products. In 2018, there were 25 serious notifications for mycotoxin contamination in cereal grains reported via the RASFF portal.6 For these reasons, we focused the method development on cereal-based food, as it is considered a high-risk commodity for mycotoxin contamination.
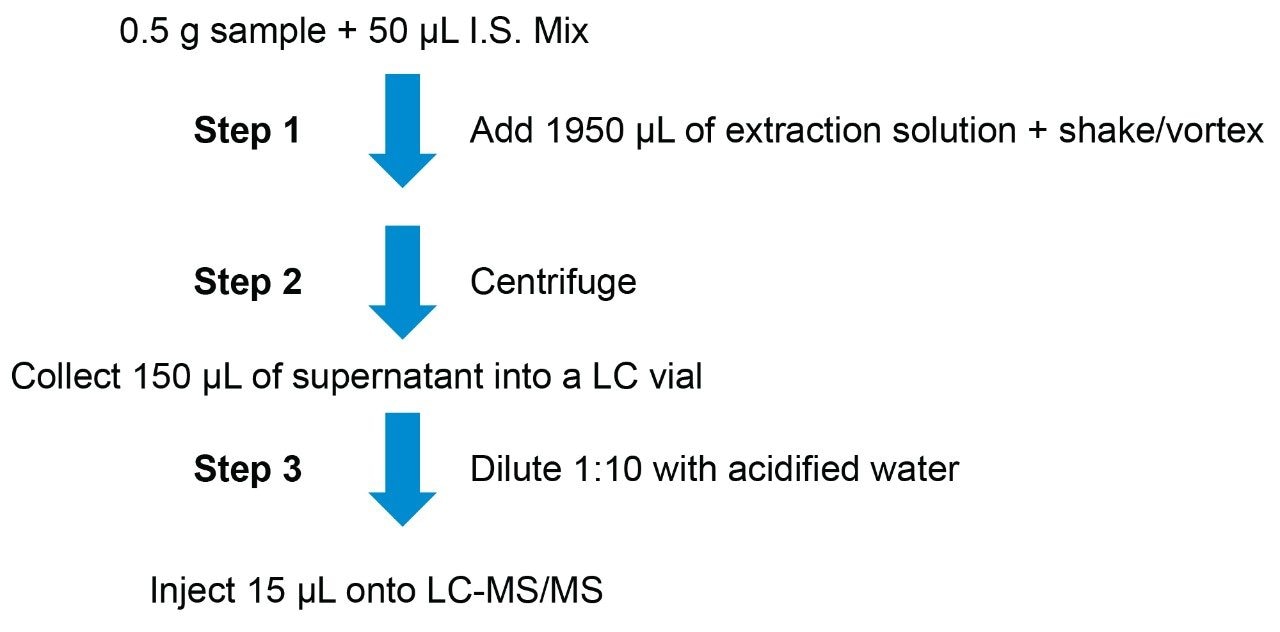
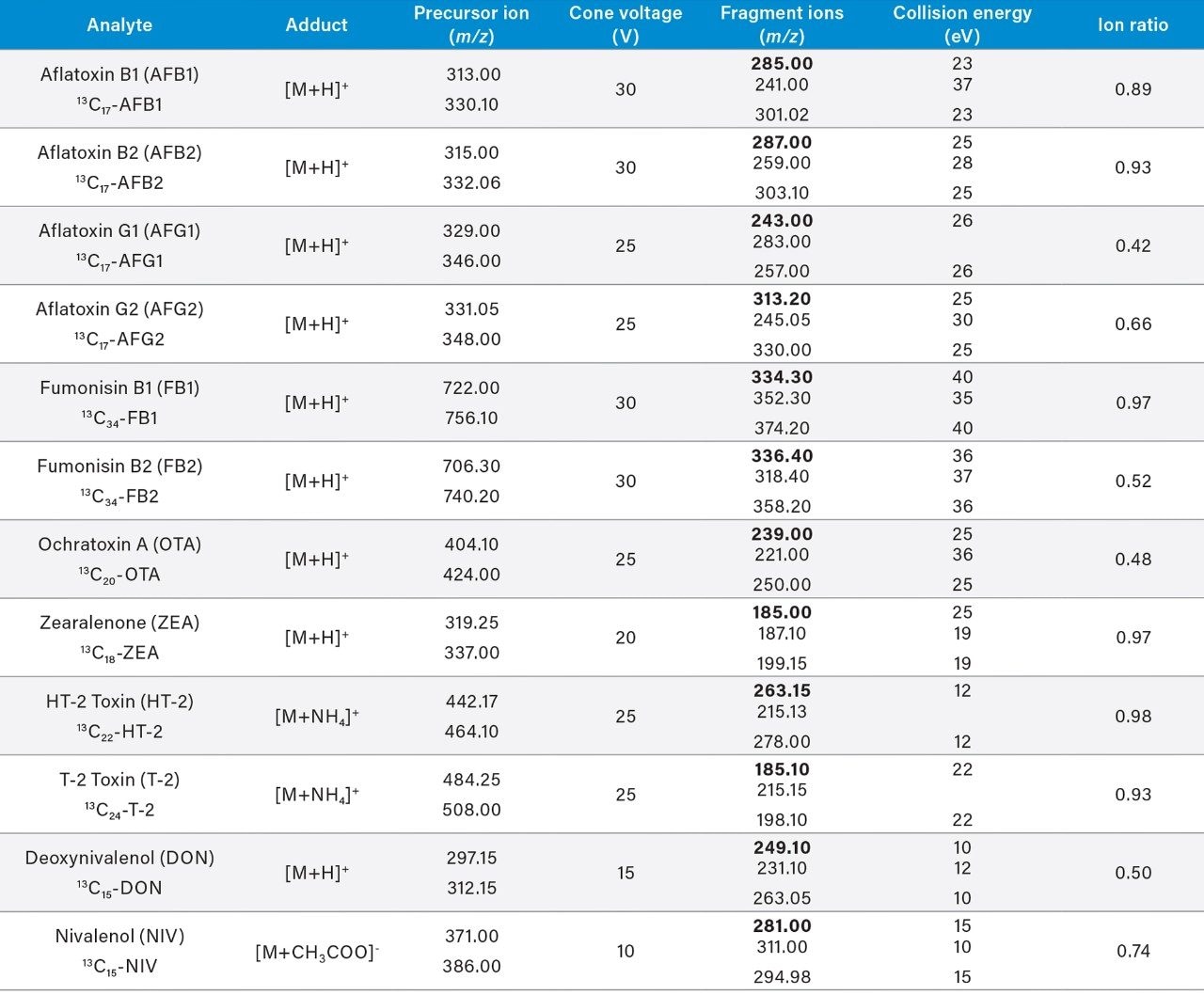
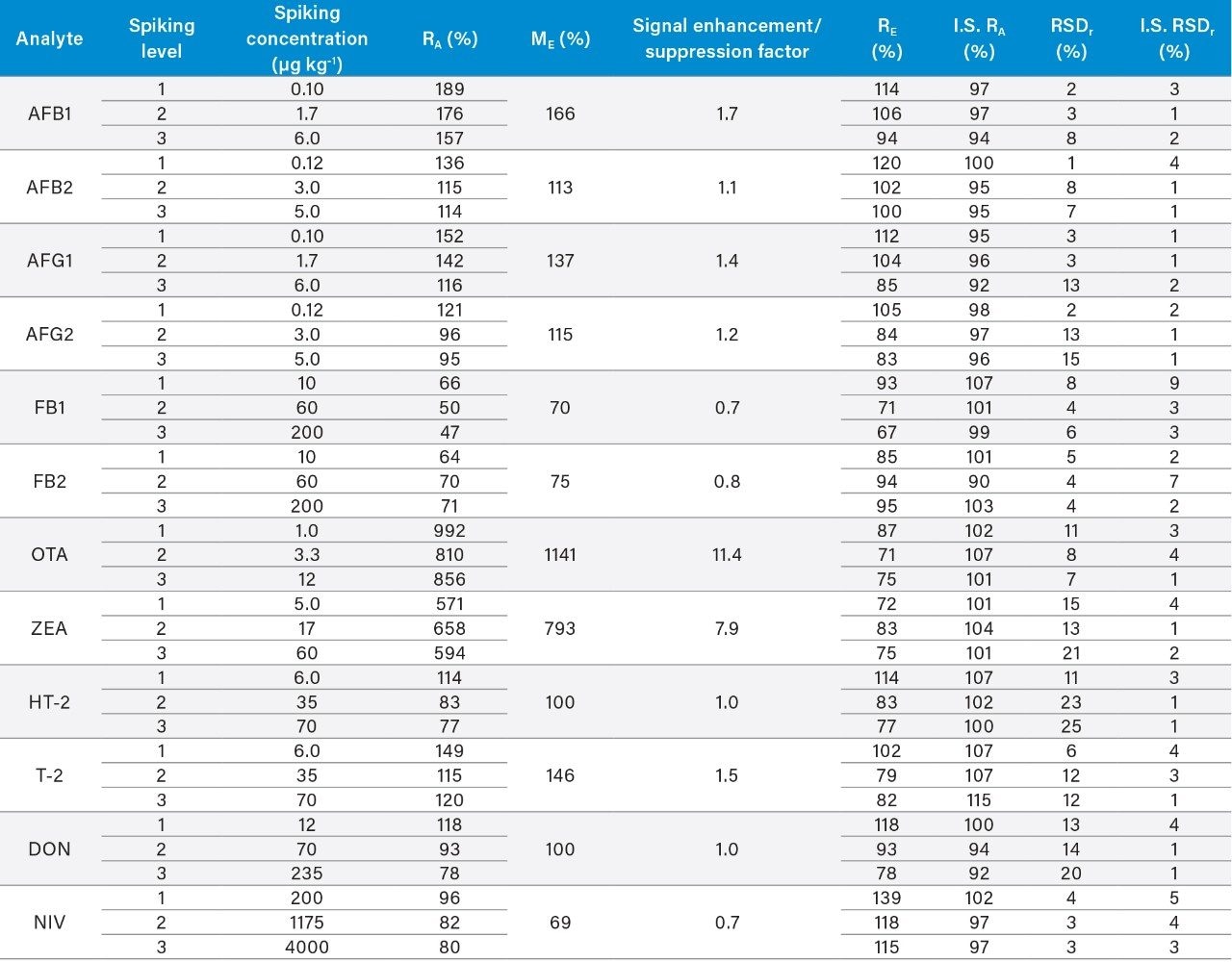
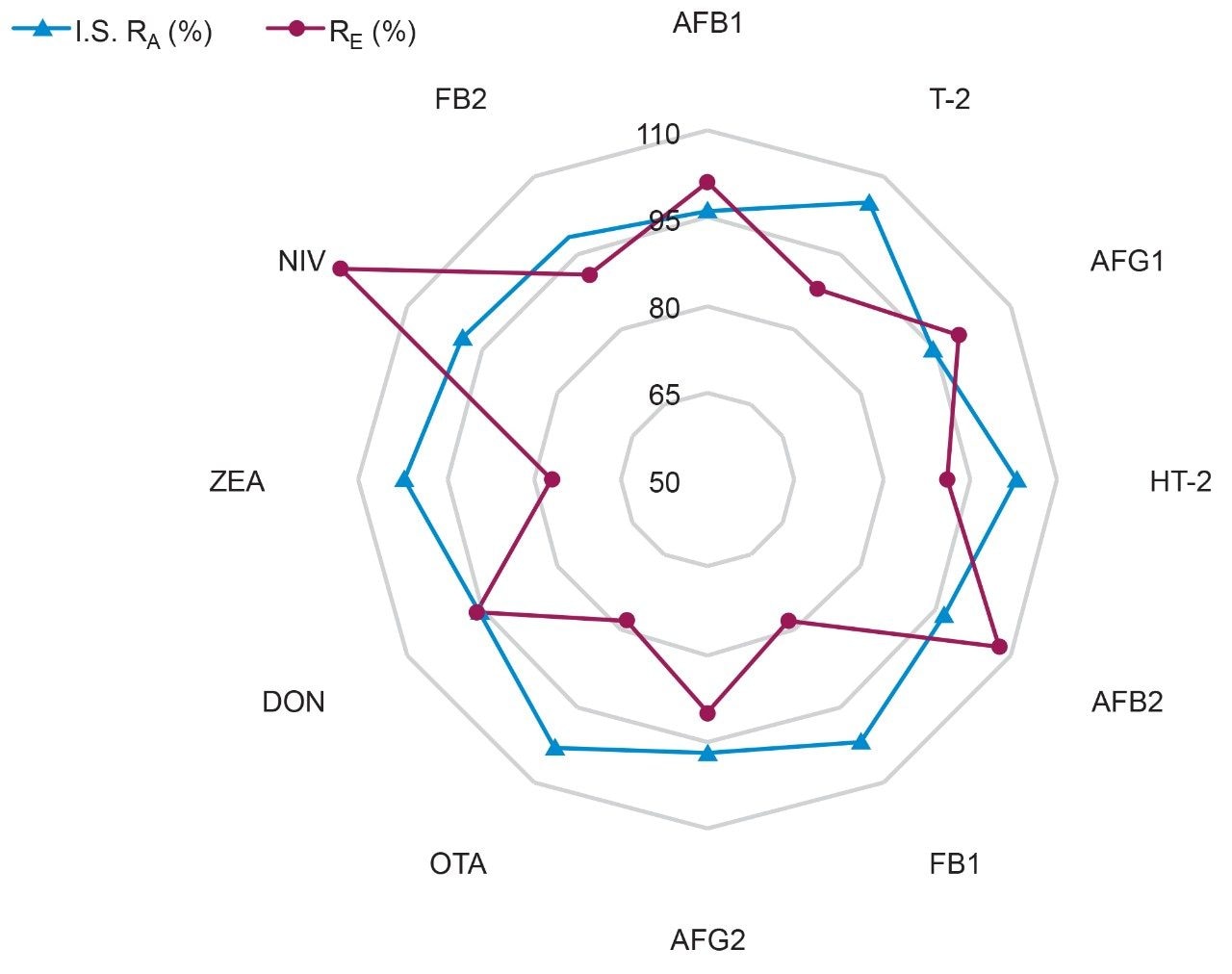
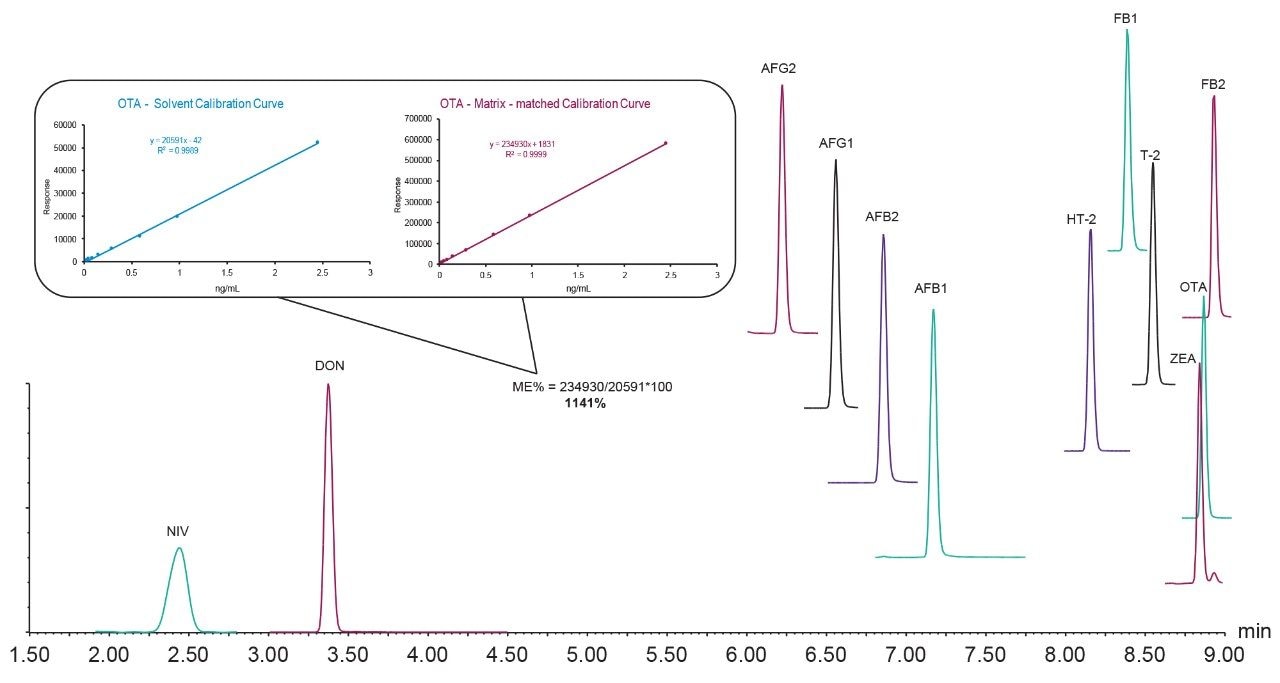
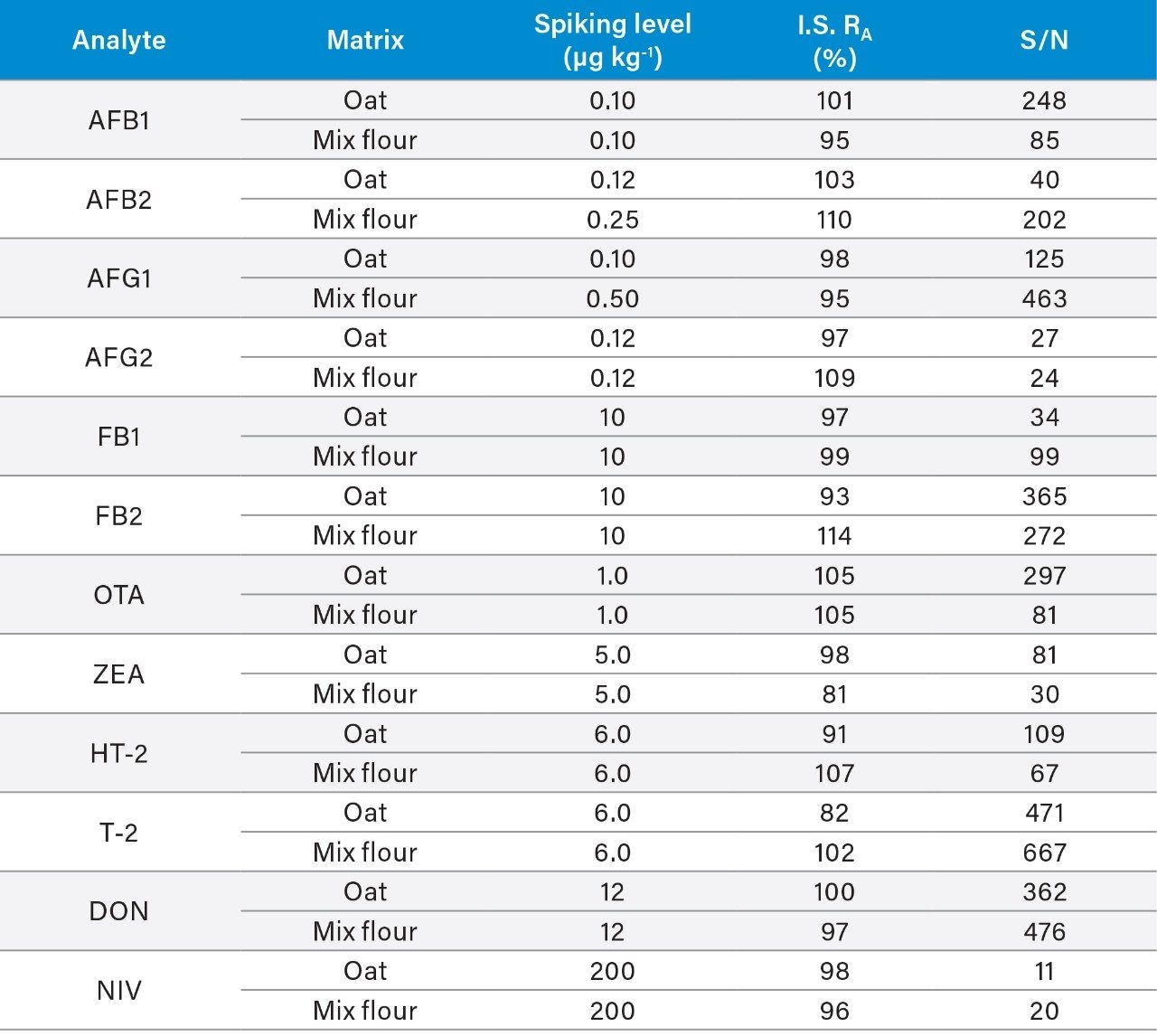
Here we describe the validation of a quantitative multi-mycotoxin method in wheat flour, which was further extended to a range of different cereal-based flours. The Waters Xevo TQ-XS Tandem Quadrupole Mass Spectrometer was chosen because it provides ultimate sensitivity and it allows the analyst to implement a simple sample preparation process, thus reducing the overall analysis time. The use of isotopically labelled internal standards was investigated to achieve better performance compared to the widely adopted external standard or standard addition methods. The analytical procedure was assessed according to criteria described by the amended European Commission Regulation No. 401/2006 and SANTE guidelines.7,8