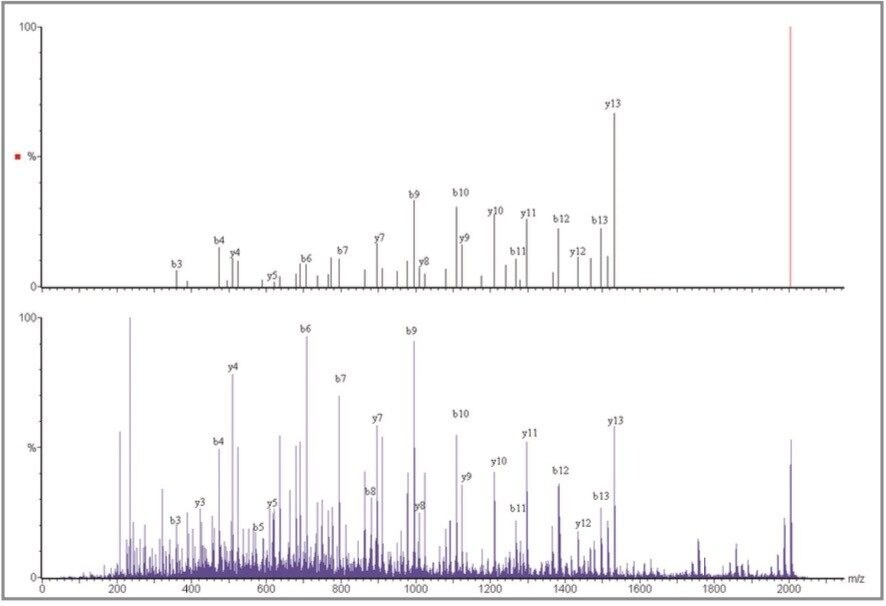
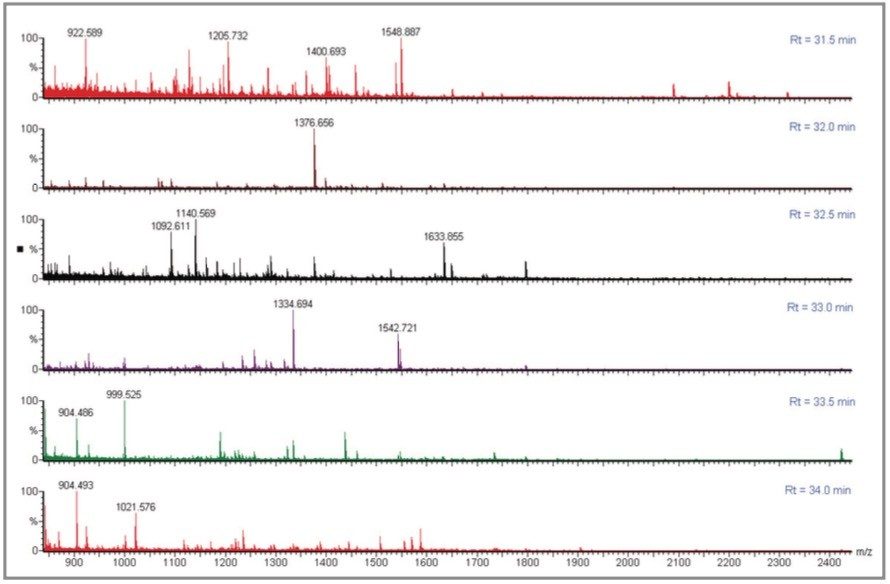
For each instrument, the fragment ion data from all 24 spots were combined into a single peaklist file, which was database-searched using MASCOT (Matrix Science).
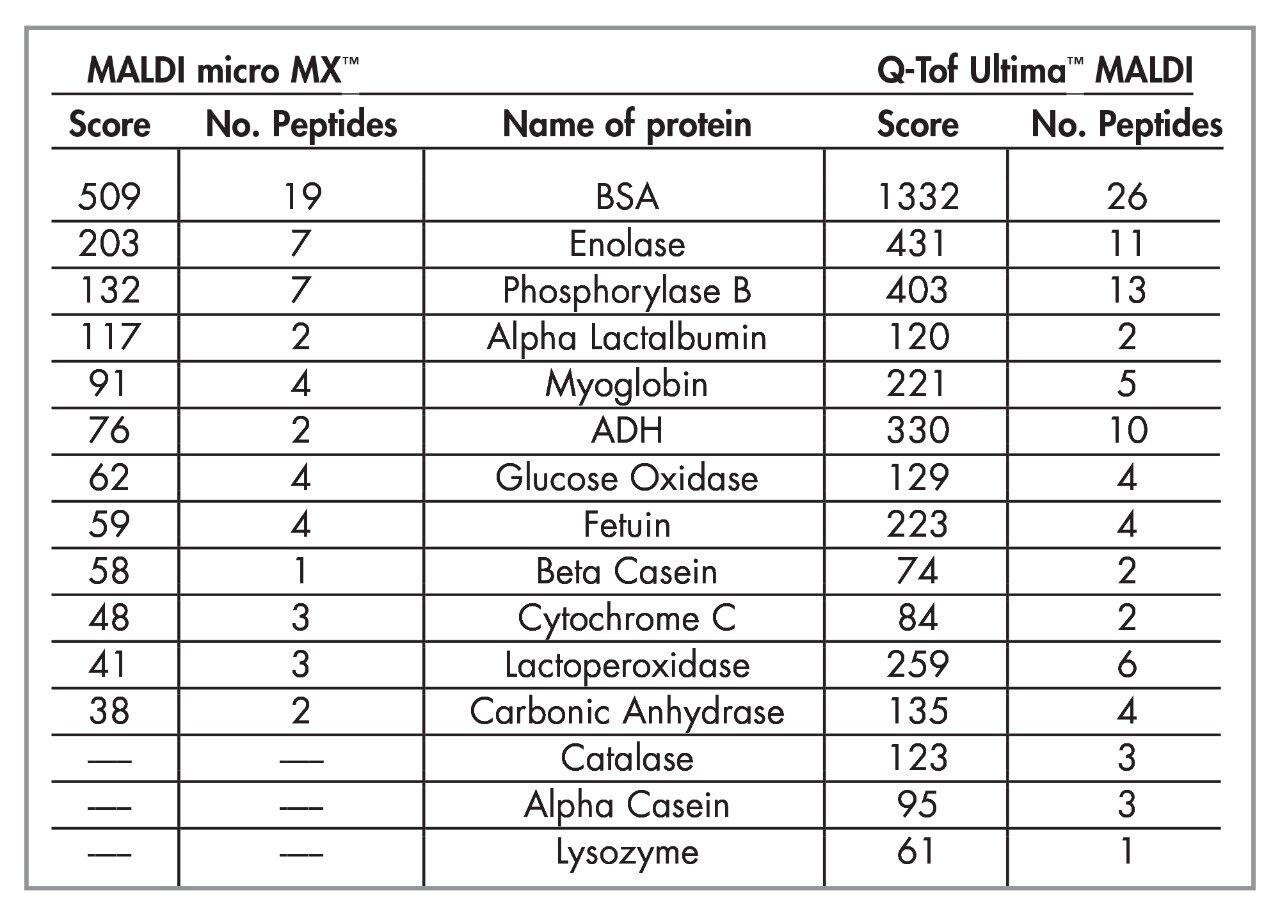
- The combination of the MALDI micro MX and the Q-Tof Ultima MALDI resulted in the identification of 14 proteins with significant confidence levels from the databank search.
- Of these 14 proteins, five proteins were common to the two techniques, while three proteins were only identified using the MALDI micro MX. An additional six proteins were only identified using the Q-Tof Ultima MALDI.
- An additional seven proteins were identified using PSD MX with a score below the positive confidence level. Of these, two were confirmed by subsequent analysis on the Q-Tof Ultima MALDI.
- In addition, four proteins were identified using Q-Tof Ultima MALDI with a score below the positive confidence level, while analysis of these by MALDI micro MX was able to confirm one of these identifications with a high score.
These results indicate that the use of MALDI on a Q-Tof geometry and parallel PSD on an axial instrument can been seen as complementary techniques. The use of both techniques provides significantly better coverage of the proteins present than either technique alone.
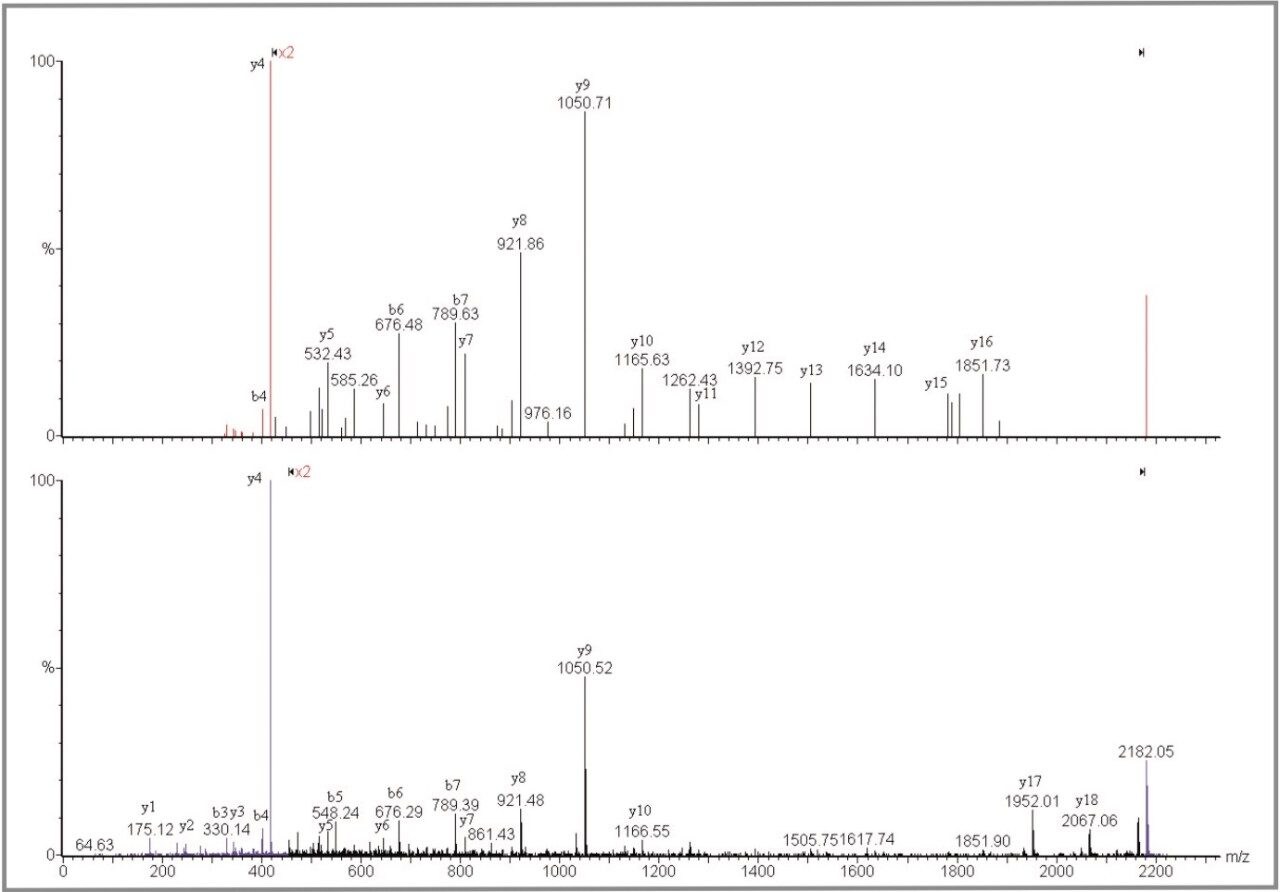
The advantage of the Q-Tof Ultima MALDI instrument is the high mass measurement accuracy. The average mass accuracy on the 21 peptides confidently identified is 2.8 ppm. The mass measurement accuracy of the Q-Tof Ultima MALDI provides a considerable improvement in the specificity of the overall experiment. This enables significantly increased confidence to be obtained from the database search, or de novo amino acid sequence to be obtained in certain cases. As can be seen in this dataset, the MS/MS fragmentation pattern from one peptide is sufficient to positively identify a protein.
In comparison, the strength of the PSD MX acquisition is the parallel nature of the experiment. This has significant benefits in terms of automating the acquisition, and greatly simplifies the experiment. For example, the protein OSMC_ECOLI has not been identified with the Q-Tof Ultima MALDI but was confidently identified in the PSD MX experiment. The precursor ions at m/z 1140.6 Da and 2155.1 Da were not automatically selected using the Q-Tof Ultima MALDI as other more intense ions were present in the spectrum, and were selected for MS/MS. In this case the parallel nature of the PSD MX experiment provides complementary information, from multiple precursor and fragment ions, which in turn enhances the specificity.