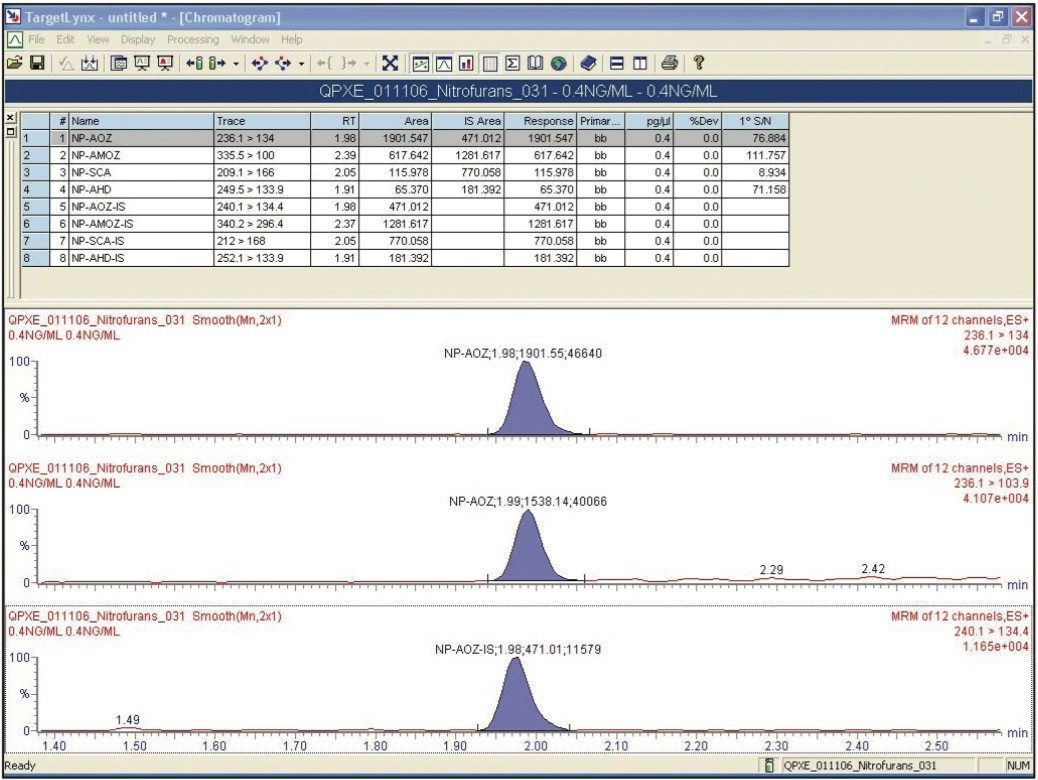
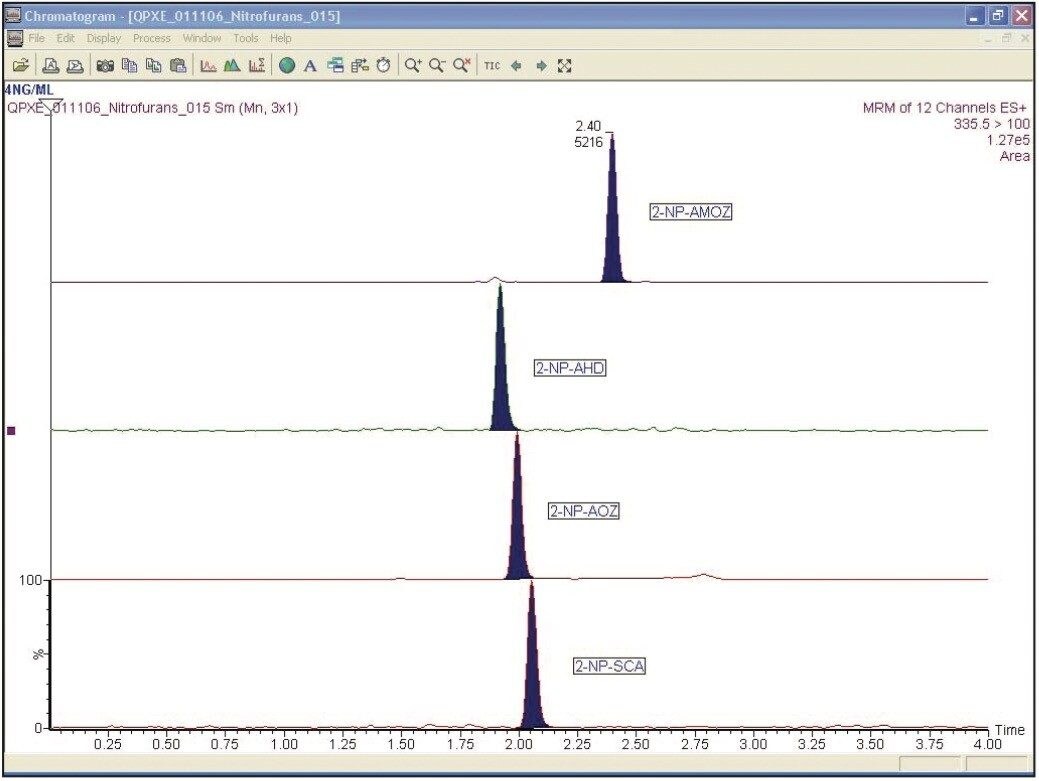
A solution containing the four derivatized metabolites and their deuterated internal standards, 2-NPAMOZ-D5, 2-NP-SCA-13C1 15N2, and 2-NP-AHD13C3 was produced at a concentration of 8 ng/mL (equivalent to 2 ppb in animal tissue) in 1:4 methanol: water (v/v). This solution was further diluted to 4, 2, 0.4, and 0.2 ng/mL (equivalent to 1, 0.5, 0.1, and 0.05 ppb in extracted samples1).
A standard curve was created for all compounds and the chromatography was assessed for each compound at each level. All concentrations reported in this technical note refer to the derivitized metabolite.
Mobile phase A was 1:4 methanol: water (v/v) + 0.5 mM ammonium acetate. Mobile phase B was 9:1 methanol: water (v/v) + 0.5 mM ammonium acetate. The column used was an ACQUITY UPLC BEH C18 1.7 μm 2.1 x 100 mm. A 50 μL injection was used with a flow rate of 0.45 mL/min.
The gradient used is described below:
0.00 min 5% mobile phase B (hold 0.2 min)
2.00 min 50% mobile phase B
2.02 min 100% mobile phase B (hold 1.1 min)
Total run time was 4.00 minutes.
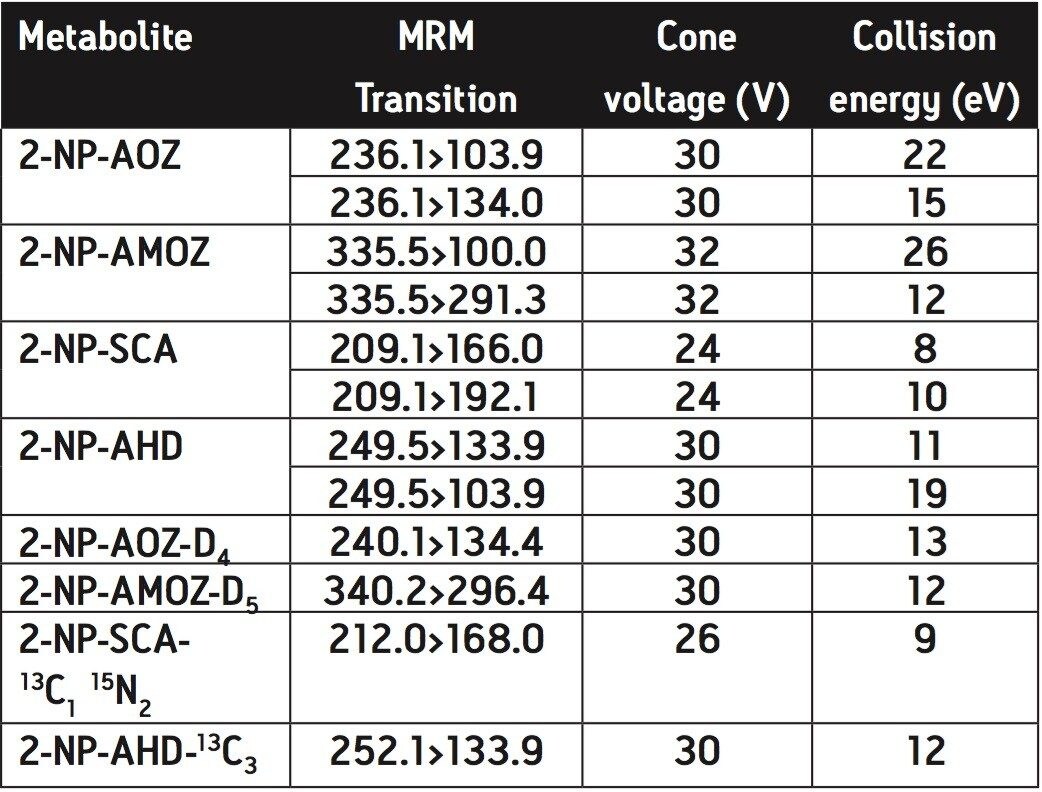
The Quattro Premier XE was set up in positive ion mode using an electrospray source. All compounds were optimized and two Multiple Reaction Monitoring (MRM) transitions were obtained for each derivatized metabolite and one for each internal standard. Two MRM transitions were monitored for each derivatized metabolite in accordance with European Union guidelines.5 The primary transition is used for quantification and the secondary transition is used for conformation purposes. These MRM transitions are listed in Table 1 along with their respective cone voltages and collision energies.