Top-Down Sequencing Using SYNAPT High Definition Mass Spectrometry
Introduction
Top-down proteomics can be described as the identification and characterization of intact proteins from Tandem Mass Spectrometry experiments, enabling the identification of Post Translational Modifications (PTMs). The top-down approach provides direct measurement of the intact mass of the protein, as well as fragment ion information relating to the amino acid sequence. This crucial information may be lost in a “bottom-up” proteomics strategy, for example, when a tryptic peptide on which a PTM resides does not ionize well. However, even with a small to medium-sized protein, the large number of ions, and the range of charge states produced during fragmentation can lead to a spectrum that can be extremely complex, challenging to interpret, and therefore, time-consuming.
The Waters® SYNAPT™ High Definition Mass Spectrometry™ (HDMS™) System, which couples high-efficiency ion mobility separations (IMS) with time-of-flight (ToF) mass analysis, provides an additional dimension of separation aiding the interpretation of these complex, multiply-charged datasets. In this application note, we separate protein mixtures by nano-scale liquid chromatography using UltraPerformance LC® (UPLC®) technology, and collect fractions for analysis. For each protein fraction, we select and fragment different charge states using the HDMS mode of analysis to separate these fragments by their differing mobility, due to their size, shape, and charge. Post-processing of these data produces simplified top-down fragment ion spectra, containing ions of mainly one charge state.
This ion mobility approach reveals low abundance fragments that would otherwise be masked by more intense species in a conventional MS/MS spectrum, and leads to enhanced sequence coverage of the protein studied. Here we present data using ubiquitin, a highly conserved small regulatory protein, separated from a protein mixture, to demonstrate the advantages of coupling ion mobility based separation with mass analysis.
Experimental
Samples
The protein mixture contained 3 pmol/µL each of ubiquitin ribonu-clease, cytochrome C, and beta lactoglobulin.
LC, Fraction Collection, and Infusion
Intact protein mixtures were separated using reversed-phase liquid chromatography. The gradient used was 20 to 40% of 0.1% formic acid in acetonitrile (solvent B) in 10 minutes, then ramped to 95% at 12 minutes. Solvent A was aqueous 0.1% formic acid. One minute wide fractions were collected. The column eluent was split inside the Triversa Nanomate (Advion BioSciences) between the LC coupler and fraction collection into a 384-well plate. 5 µL acetonitrile was added to each protein containing well and a sample volume of 1 µL was aspirated. The infused flow rate was 20 to 60 nL/min.
Mass Spectrometer
The Synapt HDMS System with Triwave™ technology, Figure 1, was used in these studies. Ions produced during electrospray ionization are sampled by a ZSpray™ source and pass through a quadrupole, which may be set to transmit a wide mass range, or to isolate ions of a specific m/z. High-efficiency ion-mobility separation is then performed in the Triwave device.
The Triwave consists of three travelling wave (T-Wave™) ion guides. The first, the Trap T-Wave, traps and accumulates ions, in this case for up to 51 msec, then these stored ions are gated into the IMS T-Wave in which the ion mobility separation occurs.
The Transfer T-Wave is then used to transport the separated ions into the orthoganol acceleration time-of-fight (oa-Tof) mass spectrometer for subsequent mass analysis. The pressure in the Trap and Transfer T-Wave regions was about 10-2 mbar of Argon and the pressure in the IMS T-Wave was 0.5 mbar of Nitrogen. Ions may be fragmented on entrance to the Trap T-Wave and/or the Transfer T-Wave. The IMS T-Wave pulse velocity and voltage may be tuned to provide the optimal ion mobility separation.
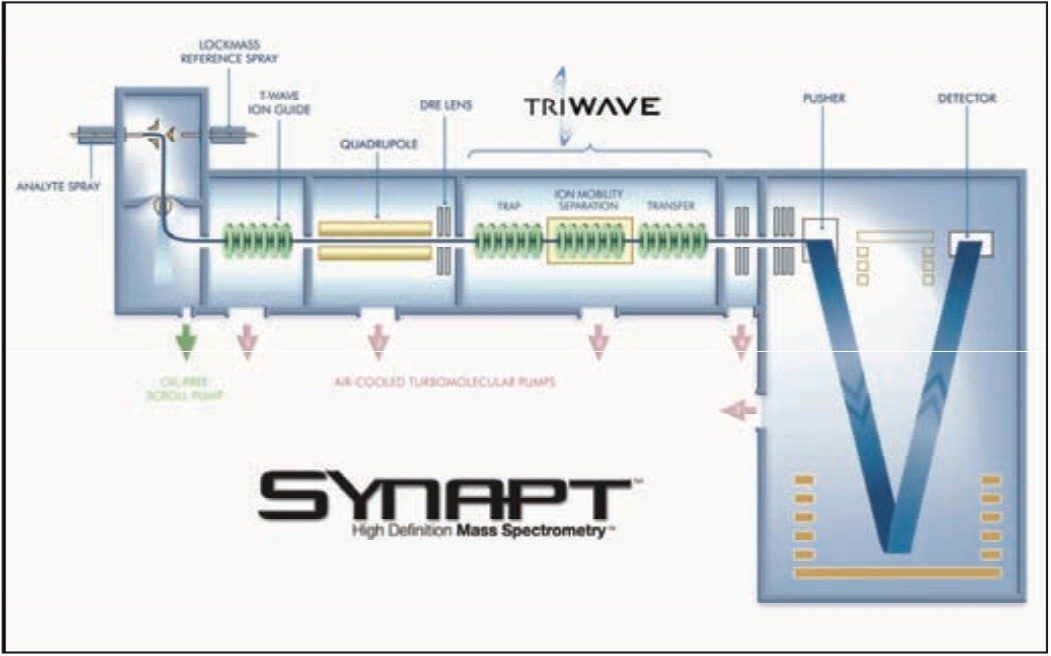 Figure 1. Schematic of the Synapt HDMS System, which incorporates Triwave technology.
Figure 1. Schematic of the Synapt HDMS System, which incorporates Triwave technology.
Results and Discussion
The five proteins were resolved chromatographically over the 15 minute separation. By combining across the individual chromatographic peaks, ToF mass spectra can be obtained for each of the intact protein species, as shown for ubiquitin in Figure 2. The multiply-charged envelope for each protein covers a wide m/z range and, using the different m/z values from the series, an accurate molecular weight of the protein can be calculated.
For ubiquitin, the calculated MW was 8565 Da. Conventional ToF MS/MS data was acquired by isolating a particular charge state of each protein in the quadrupole and fragmenting that species in the Trap T-Wave. The MS/MS spectrum obtained following the isolation and fragmentation of the 9+ ion, m/z 943 of ubiquitin is shown in the lower panel of Figure 2. It can clearly be seen that fragmentation of the highly-charged precursor ion results in a complex MS/MS spectrum, containing fragments with a variety of different charge states. This can be challenging to interpret, due to the overlapping fragments of different charge states.
The Synapt HDMS System provides the ability to reduce spectral complexity, in this case based upon separating fragment ions by their charge state. Ion mobility separations were performed whereby the MS/MS fragments were separated in the IMS T-Wave, producing data that is more easily interpreted and related to the protein sequence. Three precursor ions per protein were fragmented in the experiments and the fragmentation spectra were then used to generate sequence information.
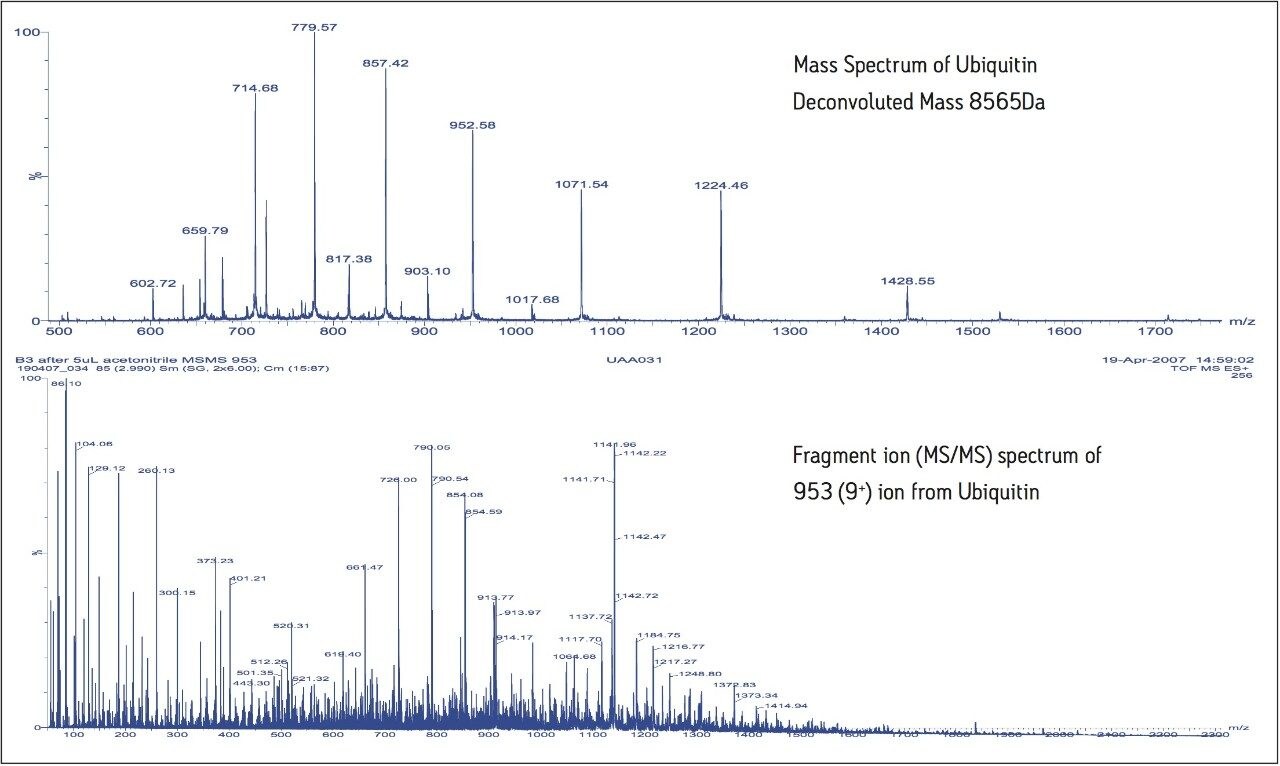 Figure 2. Upper panel shows typical ToF-MS data from LC separated ubiquitin, showing the multiply charged ion series from which the molecular weight is calculated. The lower panel shows a conventional ToF-MS/MS spectrum obtained by fragmenting the 9+ charge ion of ubiquitin, which includes ions of many different charge states.
Figure 2. Upper panel shows typical ToF-MS data from LC separated ubiquitin, showing the multiply charged ion series from which the molecular weight is calculated. The lower panel shows a conventional ToF-MS/MS spectrum obtained by fragmenting the 9+ charge ion of ubiquitin, which includes ions of many different charge states.
Data Processing
DriftScope™ Software, used to view HDMS data, displays the drift time vs. the m/z for each ion present in the data. Regions can be selected to produce new data files containing simplified spectra, mainly containing just one charge state. Maximum entropy algorithms are then used to deconvolute the spectra and fragment ion stretches, which can then be interpreted to determine the amino acidsequence of the protein.
The data presented in Figure 3 shows the DriftScope plot obtained from the Trap T-Wave fragmentation of the 9+ precursor ion from ubiquitin. The bands of color represent the intensity of the ions, with the boxed areas showing the charge state separation. The distinct separation of the singly-charged ions is typical for this type of analysis. DriftScope Software enables the user to select plot regions of interest to interrogate corresponding mass spectra. The different bands are found to correspond to different charge states or classes of ions. Selecting the entire plot produces data equivalent to that acquired without ion mobility separation, and hence allows a comparison of data produced by conventional MS/MS with ion mobility separated MS/MS (HDMS).
A comparison is shown in Figure 4 from the fragmentation of the m/z 952.5 ion from ubiquitin. Selection of the quadruply charged (4+) region from the analysis in HDMS mode produces an increase in the signal-to-noise level of the spectrum when compared to the conventional MS/MS data. This allows clear assignment of amino acid sequence to the fragment ions, and provides increased sequence coverage.
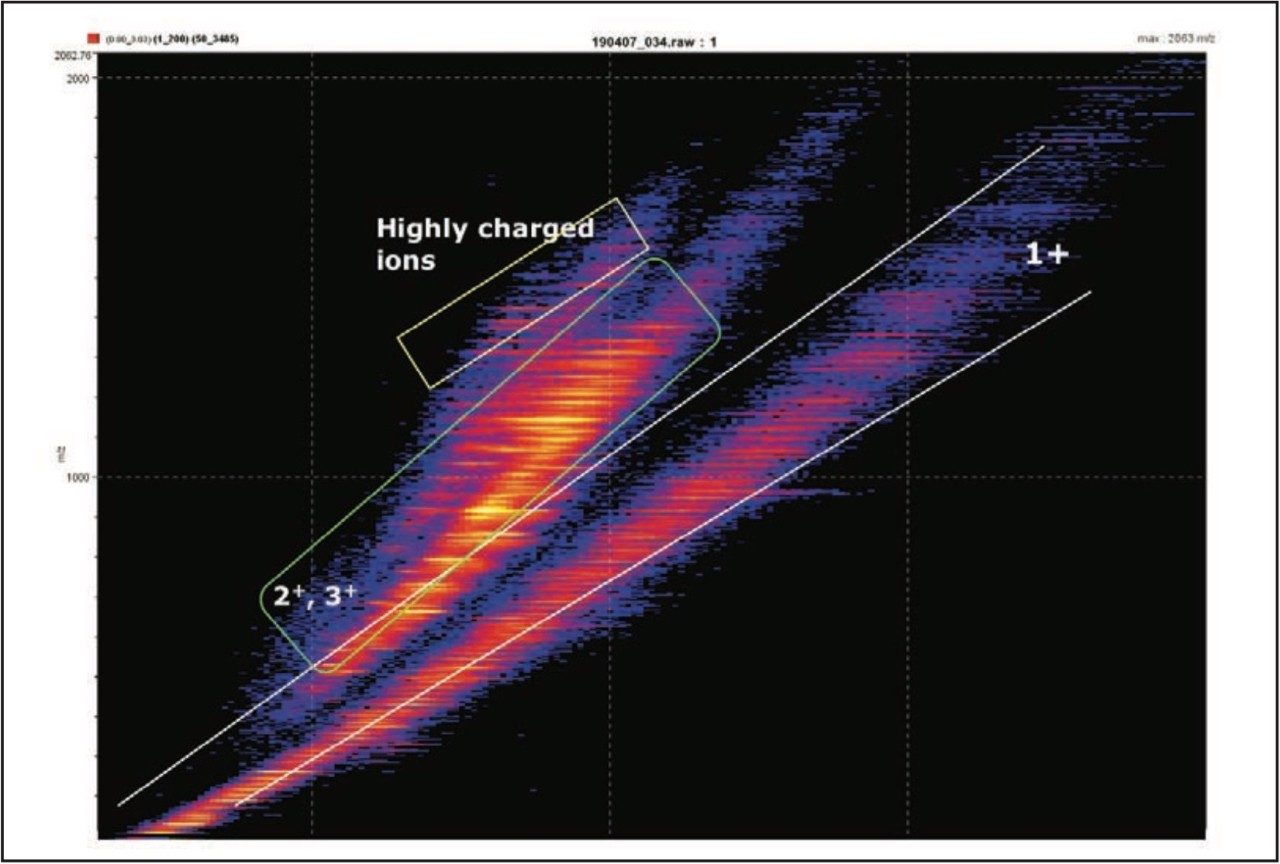 Figure 3. Drift time plots vs. m/z for the Trap T-wave fragmentation of one of the multiply charged precursor ions from ubiquitin, showing separation into distinct charge state bands.
Figure 3. Drift time plots vs. m/z for the Trap T-wave fragmentation of one of the multiply charged precursor ions from ubiquitin, showing separation into distinct charge state bands.
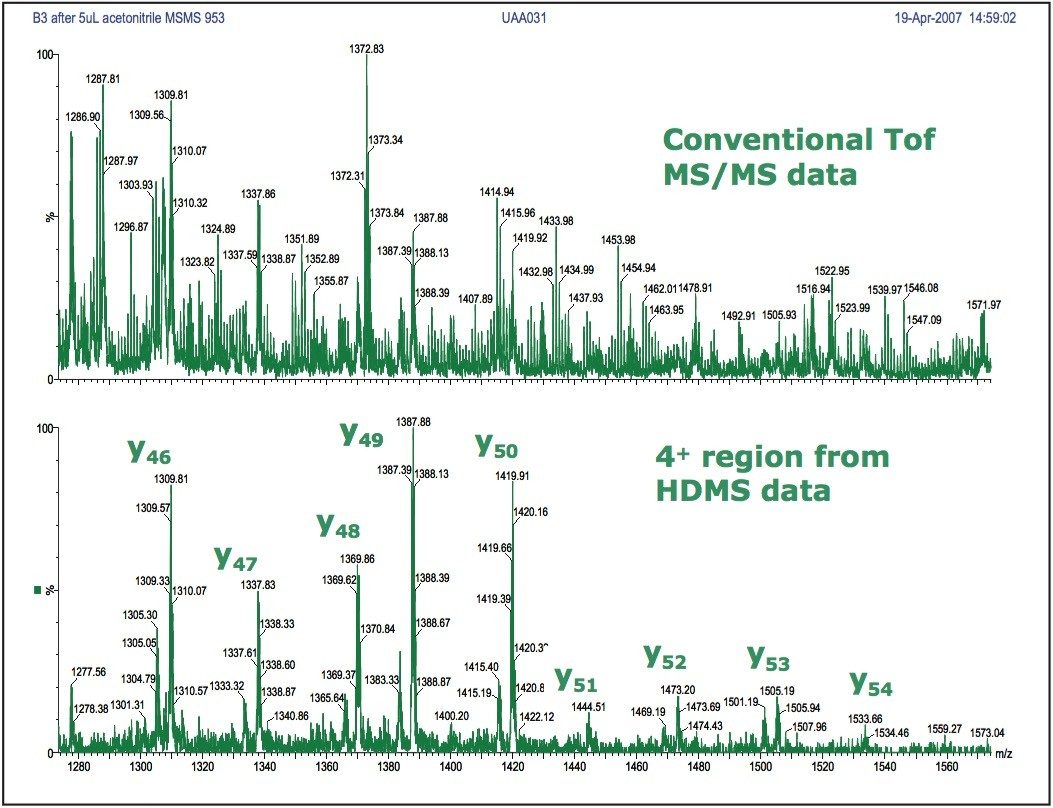 Figure 4. Improved signal-to-noise level observed with HDMS mode for the quadruply charged y’’ series fragment ions from ubiquitin.
Figure 4. Improved signal-to-noise level observed with HDMS mode for the quadruply charged y’’ series fragment ions from ubiquitin.
Figure 5 shows data selected from the singly-charged region of the HDMS data. It is interesting to note that the singly-charged b11 ion observed in the HDMS mode is masked by the more intense y44 4+ ion in the conventional ToF-MS/MS data. In all cases, examination of the ion mobility separated fragments reveals information that was not observed, or was hidden by more intense species in the conventional ToF-MS/MS acquisition.
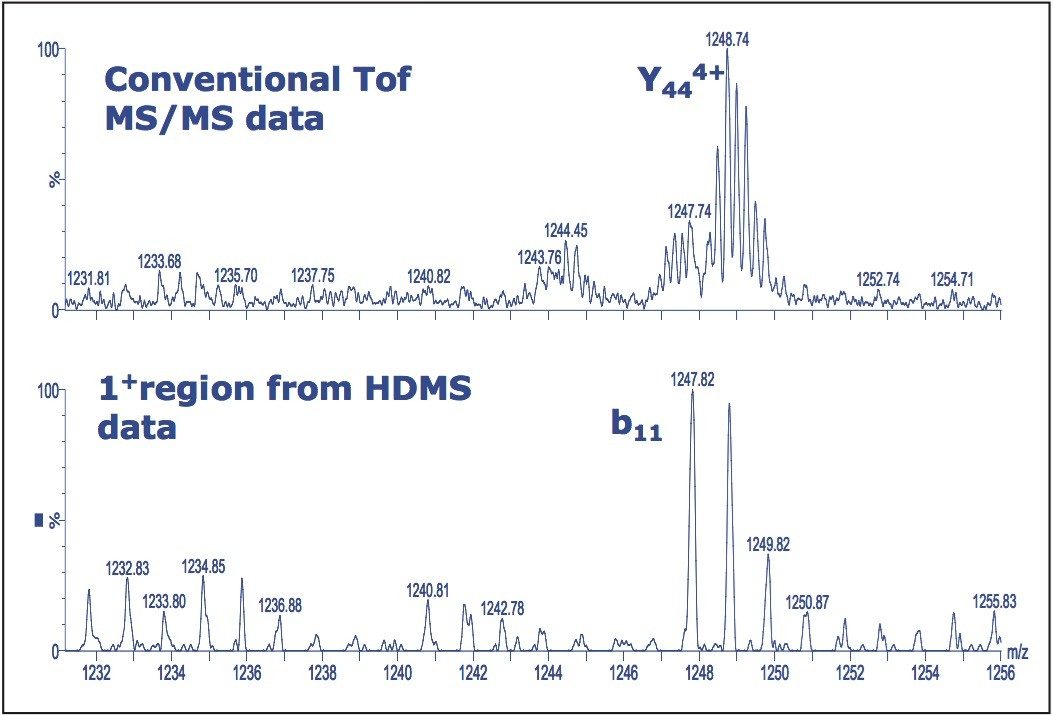 Figure 5. Selection of the 1+ region reveals the b11 ion, lower trace, which is masked by the more intense y444+ ion in the conventional ToF-MS/MS data shown in the upper trace.
Figure 5. Selection of the 1+ region reveals the b11 ion, lower trace, which is masked by the more intense y444+ ion in the conventional ToF-MS/MS data shown in the upper trace.
Sequence Coverage for Proteins of Different Molecular Weight
The sequence information (b and y’’ ions) obtained from the four different proteins analysed in ToF-MS and HDMS mode was investigated. This information is summarized in Figure 6, where the percentage increase in sequence coverage for the HDMS data is plotted for each of the proteins. Sequence coverage was increased in all experiments where ion mobility was employed as an additional separation dimension.
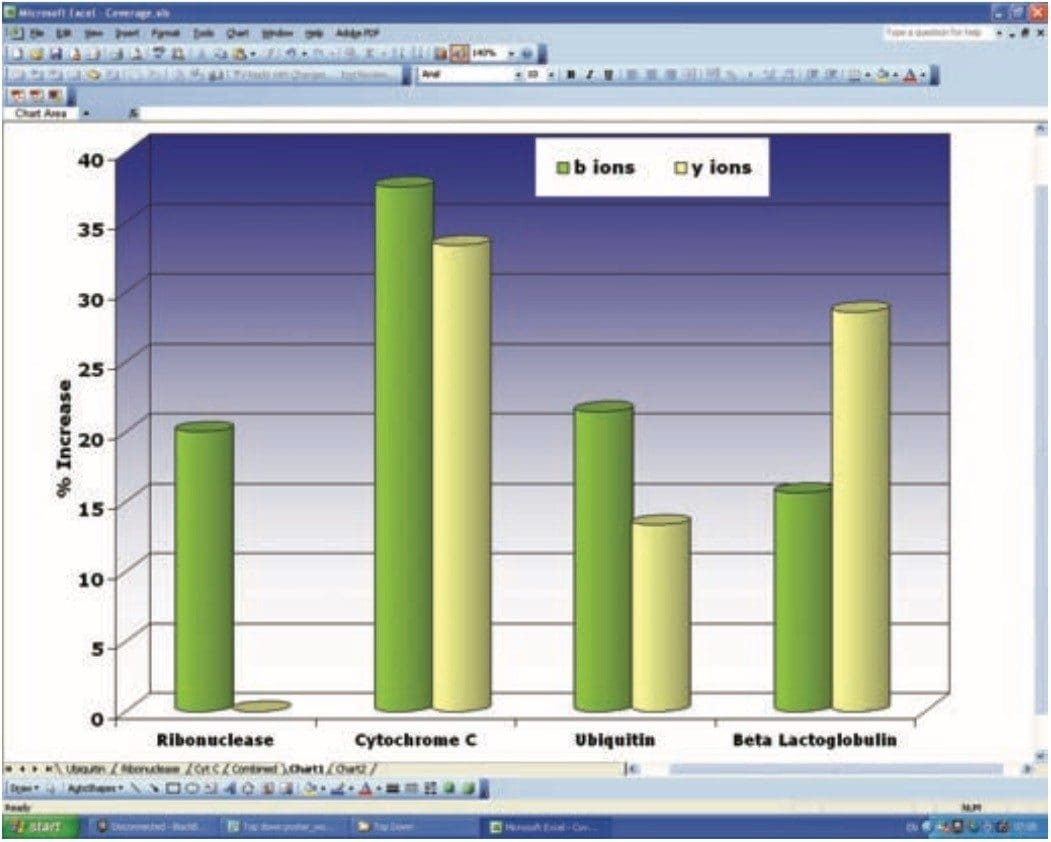 Figure 6. Percentage increase in b and y ions when HDMS data is compared with conventional ToF-MS/MS data.
Figure 6. Percentage increase in b and y ions when HDMS data is compared with conventional ToF-MS/MS data.
Conclusion
- For the small to medium sized proteins analyzed, the HDMS data produces an increase in sequence coverage resulting in more complete protein characterization compared to the conventional ToF-MS/MS experiment
- Simplified mass spectra are generated in the HDMS analysis where fragment ions, produced from highly charged precursor ions in the TRAP T-Wave, are separated in the IMS T-Wave
- Low intensity species which are not observed in a conventional ToF-MS/MS analysis could be identified and characterized
The travelling wave device described here is similar to that described by Kirchner in US Patent 5,206,506 (1993).
Featured Products
720002342, September 2007