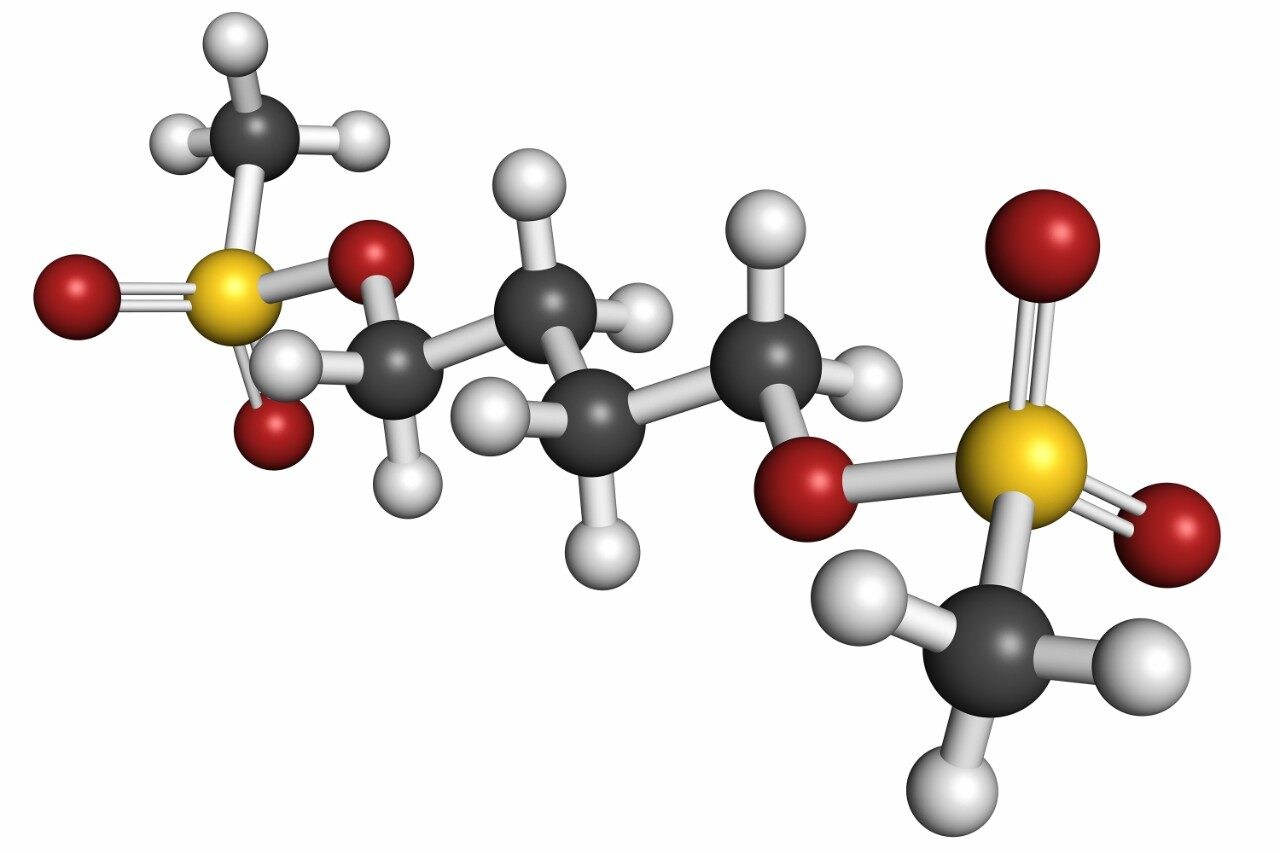
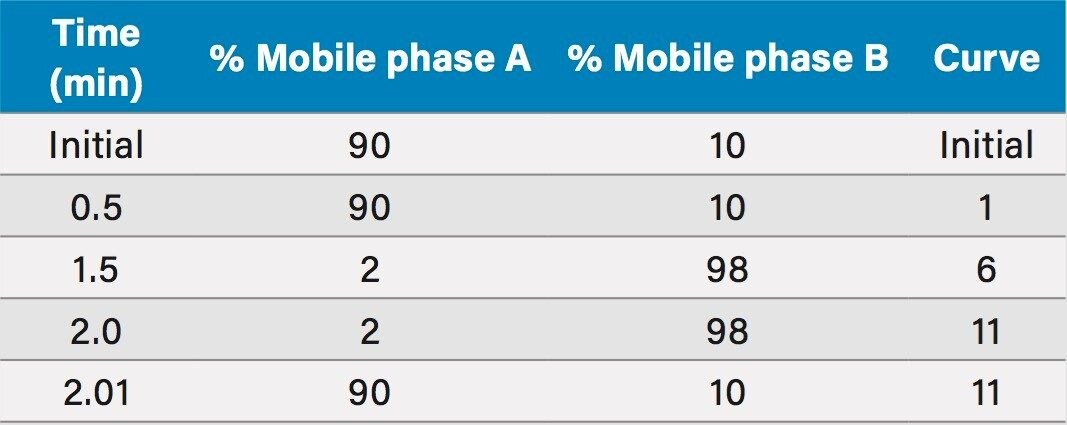
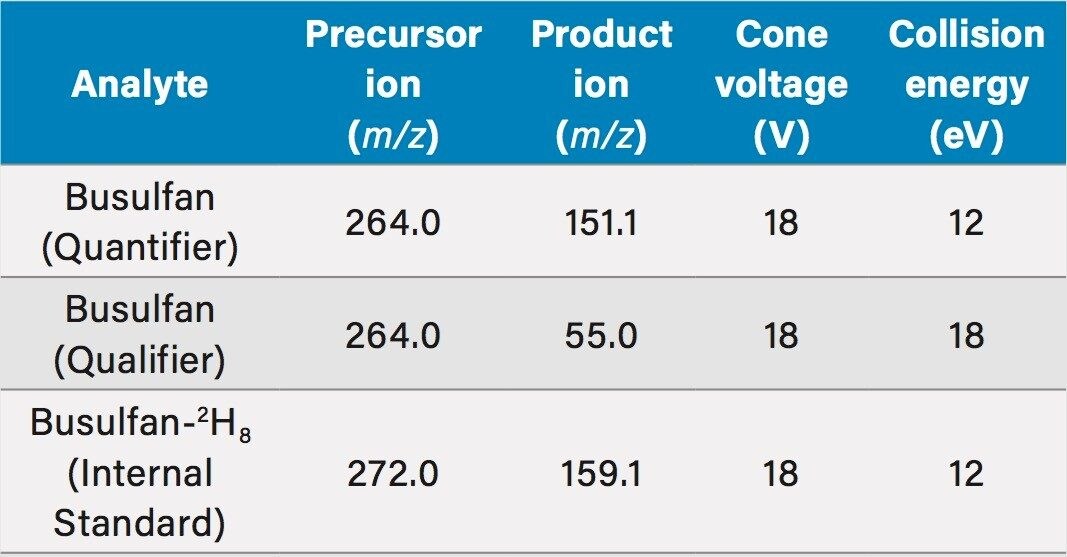
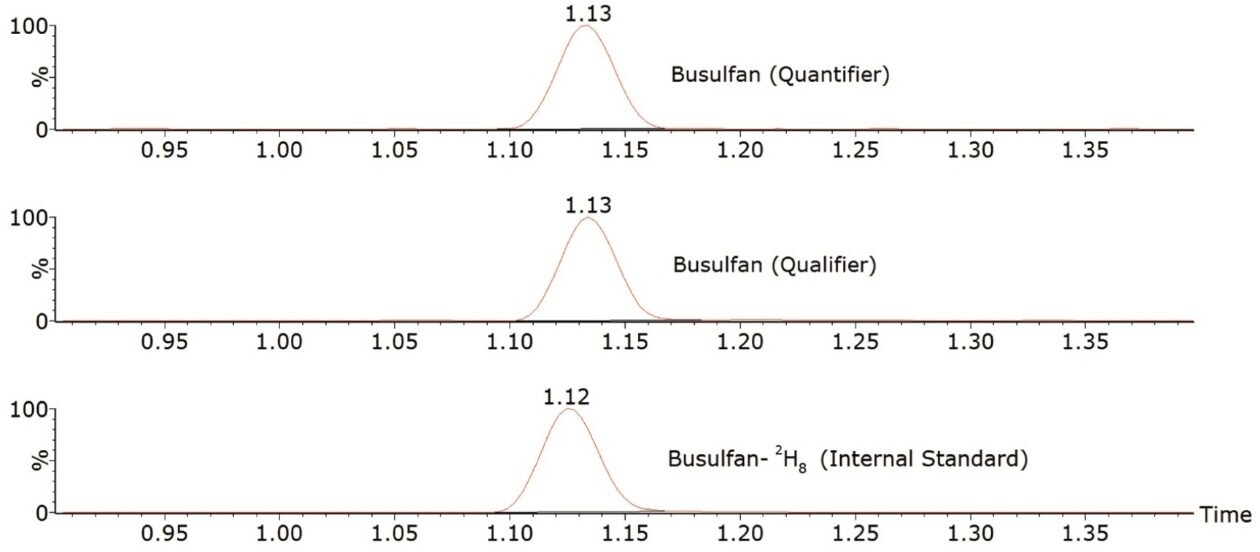
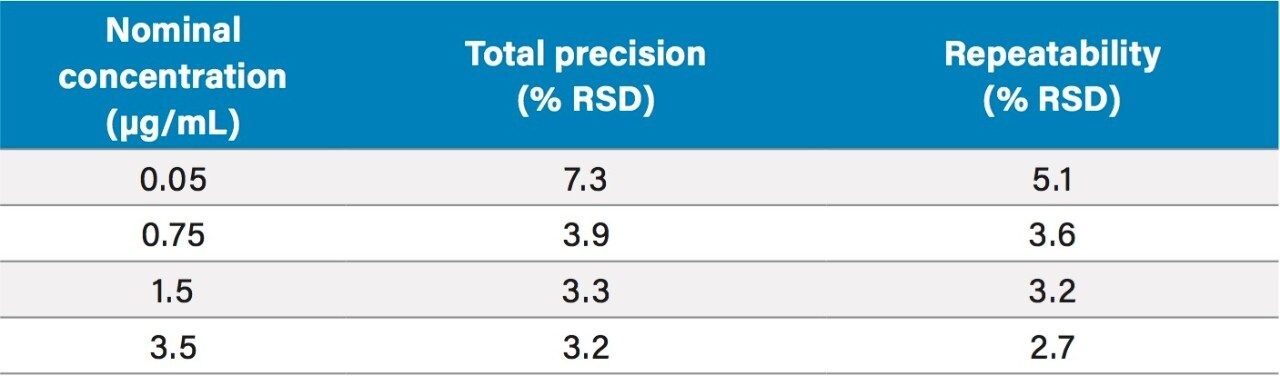
The method was shown to be linear over the range of 0.0175–6.51 µg/mL when different ratios of high and low concentration pools of busulfan were combined and analysed.
Matrix effects were evaluated at low (0.05 µg/mL) and high (3.5 µg/mL) busulfan concentrations in plasma (n=6). The matrix factor range was 0.92 to 1.06 (mean 0.99) for low concentrations compared with 0.99 to 1.12 (mean 1.08) for high concentrations. Use of busulfan internal standard adjusted response compensated for this enhancement with matrix factor ranges of 0.98 to 1.02 (mean 0.99) and 0.95 to 1.00 (mean 0.98) for low and high concentrations, respectively.
Potential interference from endogenous compounds (albumin, bilirubin, cholesterol, triglycerides, and uric acid) and the exogenous material intralipid (20% emulsion) spiked at high concentrations was assessed by determining the recovery of busulfan (n=3) from low and high pooled plasma samples (0.05 µg/mL and 3.5 µg/mL). Recovery ranged from 85.1–106.1%. Recovery ranged from 93.2–103.0% when assessing the effects of the presence of acetaminophen, fluconazole, ketoconazole, itraconazole, phenytoin, posaconazole, and voriconazole. A substance was deemed to interfere if a recovery range of 85%–115% was exceeded.
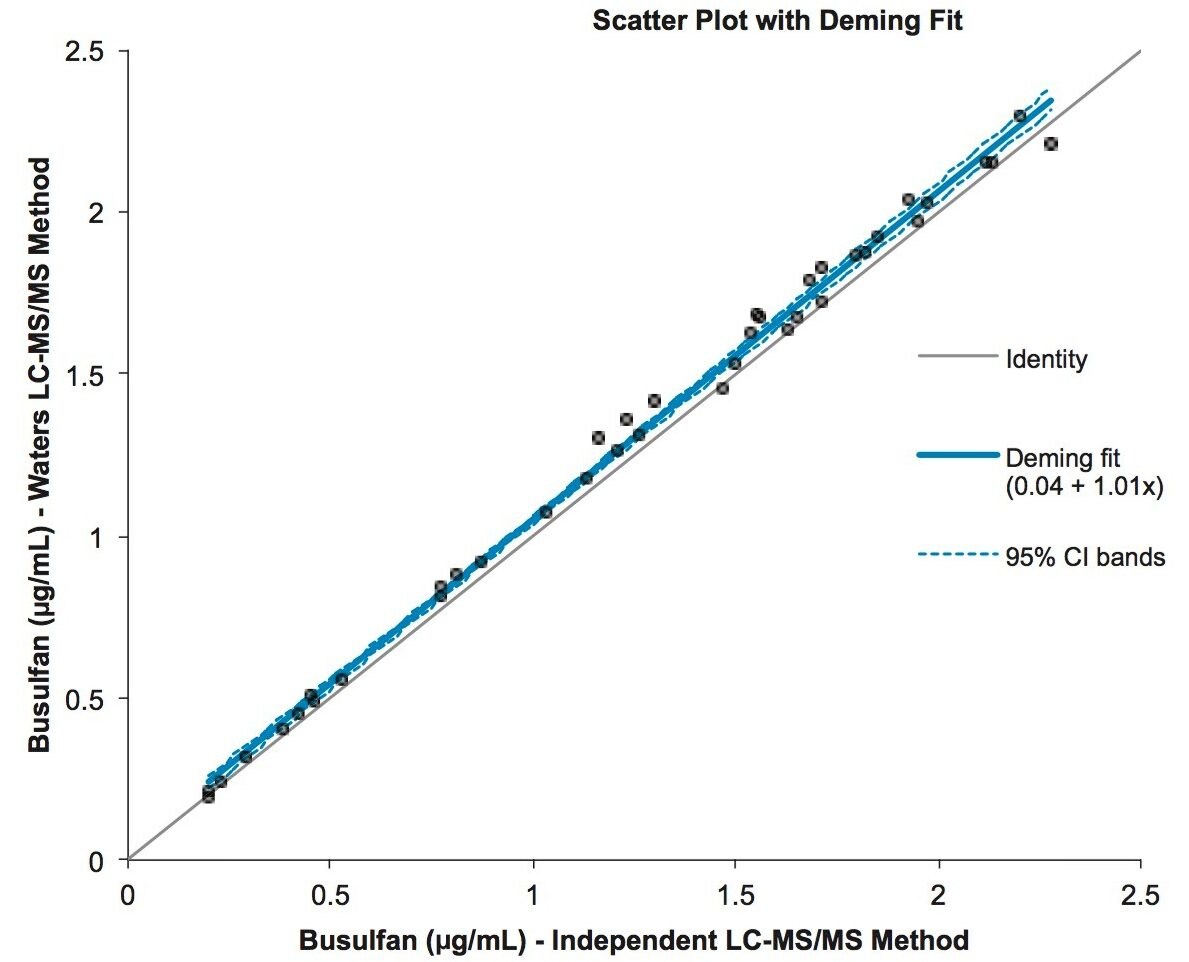
Good correlation between plasma sample concentrations and an independent, validated, LC-MS/MS method was achieved as described by the Deming equation y = 1.01x + 0.04 (n=40, range 0.02–2.28 µg/mL, Figure 3).